Сколько весит автомобиль «Ока» (ВАЗ
Вес снаряженного и заправленного автомобиля «Ока» составляет 645 кг, а его полная масса – 975 кг.
«Ока» (ВАЗ – 1111) является малогабаритным и малолитражным автомобилем, впервые разработанным и выпущенным на Волжском автомобильном заводе в 80-х годах. Цель разработчиков – создание доступного, востребованного и экономичного средства передвижения. Сколько весит «Ока»? Полная масса автомобиля определяется снаряженной массой и массой полезного груза. Итак, сегодня мы узнаем об особенностях, и основных характеристиках автомобиля «Ока».
Автомобиль «Ока»: исторические факты
Вес снаряженного и заправленного автомобиля «Ока» составляет 645 кг, а его полная масса – 975 кг.
Прототипом «Оки» стал японский Daihatsu Cuore 1985 года выпуска, у которого разработчики «позаимствовали» основные элементы дизайна кузова и ряд технических решений. Однако ходовая часть и силовой агрегат отечественного автомобиля были разработаны заново. Правда, вместо 3-цилиндрового двигателя, оказавшегося не готовым к началу выпуска автомобиля, было решено оснастить «Оку» 2-цилиндровым мотором.
Правда, вместо 3-цилиндрового двигателя, оказавшегося не готовым к началу выпуска автомобиля, было решено оснастить «Оку» 2-цилиндровым мотором.
Согласно проекта, «Ока» стала настоящим «народным» автомобилем, а ее модификации поражали многообразием: базовая модель с двухцилиндровым двигателем, социальная версия для инвалидов, модель с 2-цилиндровым двигателем ВАЗ-11113 (объем 0,75 л), мелкосерийная спортивная версия с четырехточечными ремнями безопасности.
Дефолт и девальвация рубля в 1998 году принесли небывалую популярность автомобилю «Ока» — благодаря резкому падению цены на данное средство передвижения. Так что вплоть до 2005 года заводами-производителями активно наращивался выпуск модели сегмента А. В дальнейшем внутренний и внешний рынок постепенно стал заполняться современными китайскими иномарками, которые значительно «подвинули» отечественную «Оку». К тому же, адаптация двигателя автомобиля под нормы Евро-2 оказалась нерентабельной и в 2006 году его производство было остановлено.
Автомобиль «Ока» — вес и другие технические характеристики
| Машина характеризуется: | Показатель |
| скоростью (максимальной) | 130 км/ч |
| расходом топлива (на 100 км) | 4,3 л |
| объемом бензобака | 30 л |
| временем разгона до 100 км/ч | 24 с |
| массой «Оки» (снаряженной/полной) | 645/975 кг |
| размером шин | 135/80 R12 |
| объемом двигателя | 750 см3 |
| расположением двигателя | поперечное, впереди |
| количеством оборотов | 5600 |
| крутящим моментом | 52/3200 н*м |
| системой питания | карбюратор |
| газораспределительным механизмом | ОНС |
| цилиндрами (количество/расположение/диаметр/количество клапанов на цилиндр) | 2/рядное/82 мм/2 |
| ходом поршня | 71 мм |
| степенью сжатия | 9,6 |
| видом топлива | АИ-92 |
| передними тормозами | дисковые |
| задними тормозами | барабанные |
| типом рулевого управления | шестерня-рейка |
| приводом | передний |
| количеством передач | механическая коробка — 4 |
| подвеской (передней/задней) | амортизационная стойка/винтовая пружина |
| типом кузова | хэтчбек |
| количество дверей/мест | 3/4 |
| клиренсом | 150 мм |
| объемом багажника (максимальный/минимальный) | 360/210 л |
| годом выпуска | 1996 |
Какие характеристики машины обычно интересуют покупателя? Как правило, это расход топлива, скорость, цена и другие показатели. О том, сколько весит «Ока», многие задумываются далеко не в первую очередь. Однако следует помнить, что существует прямая взаимосвязь между массой авто и остальными его характеристиками.
О том, сколько весит «Ока», многие задумываются далеко не в первую очередь. Однако следует помнить, что существует прямая взаимосвязь между массой авто и остальными его характеристиками.
К примеру, более тяжеловесная машина должна быть оснащена более мощным двигателем – для развития необходимой скорости. В свою очередь, для «внушительного» транспортного средства потребуется больше топлива, а значит, эта статья расходов также увеличится. Подобные «гигантские» модели были популярны в 50-60-х годах прошлого века – масса отдельных «экземпляров» достигала трех тонн!
Правда, современные автомобили обладают намного меньшими весовыми «категориями» и выигрывают за счет усовершенствования других важных характеристик.
Сколько весит холодильник «Ока-6М»?
В советское время под маркой «Ока» выпускали не только автомобили, но и двухкамерные холодильники. Наиболее популярная модель тех времен – «Ока-6М», отличительной особенностью которой являлся ободок черного цвета, обрамлявший передние дверцы. Полоски такого же цвета разделяли морозильную и холодильную камеры и служили своеобразным «товарным знаком» изделия.
Полоски такого же цвета разделяли морозильную и холодильную камеры и служили своеобразным «товарным знаком» изделия.
Холодильник общим объемом 300 литров прекрасно вмещал запас домашней провизии. Правда, металлические решетчатые полки были немного неудобны для хранения мелких продуктов, а пролитая жидкость сразу стекала на нижние отделения холодильника. В отличие от современных «рефрижераторов», советская «Ока» была достаточно шумным агрегатом – особенно ощутимо это было в ночное время.
Теперь мы знаем, сколько весит «Ока», а также другие занимательные факты из «жизни» автомобилей и холодильников этой марки.
Грузовики марки «Ока»
С. Цыганов, фото автора и М. Шелепенкова
Первые опыты по созданию грузопассажирской модификации «Оки» относятся к девяностым годам. Тогда малолитражку делали на трех заводах, но неформальное первенство принадлежало челнинскому ЗМА. В Набережных Челнах появилось множество тюнинговых фирмочек, предлагавших для «Оки» варианты внешнего обвеса, модернизированные салоны, оригинальное допоборудование. Известны и более серьезные проекты, в которых тюнинговые фирмы выступали в кооперации с заводом, – «Динара», «ОКА-LADY», «Алсу», «Ока-кабриолет».
Известны и более серьезные проекты, в которых тюнинговые фирмы выступали в кооперации с заводом, – «Динара», «ОКА-LADY», «Алсу», «Ока-кабриолет». С июля «Ока» выпускается с новым обвесом тольятинской фирмы «Юрол»
Из-за небольшого спроса производство этих машин колебалось от единиц до десятков экземпляров (максимум), изготовленных для конкретных заказчиков. исключением является проект «Астро11301» – это «Ока» с четырехцилиндровым МеМЗ-245, но и она на фоне общего выпуска осталась мелкосерийной модификацией.
С транспортом для перевозки грузов в Челнах экспериментировали много, иногда даже удачно. В 2000 г. на Автосалоне показали рамный пикап ЗМА-1901 «Ока-Гном» на базе… автомобильного прицепа (!) 8125, оснащенный маленьким сельскохозяйственным дизелем. Это был в чистом виде артефакт, оставшийся в единственном экземпляре, но фургоны на базе малолитражки производились в товарном количестве. Модификация ВАЗ-11113 «Тойма» (фургон с пластиковым кузовом-надстройкой и двумя распашными дверьми сзади) с 2000 г. разошлась в нескольких сотнях экземпляров.
Модификация ВАЗ-11113 «Тойма» (фургон с пластиковым кузовом-надстройкой и двумя распашными дверьми сзади) с 2000 г. разошлась в нескольких сотнях экземпляров.
Серпуховский автозавод до времени оставался в арьергарде экспериментов со стайлингом и новыми модификациями. Лишь на рубеже тысячелетий появился проект коммерческого универсала – «Ока» без заднего остекления. На базе этой машины даже предложили модель для МЧС, со штаным спасательным оборудованием и в соответствующей раскраске.
| На площадке готовой продукции много машин не скапливается – раскупают |
А еще сделали настоящую грузовую машину, по компоновке похожую на сельскохозяйственный трактор – с кузовом впереди кабины и приводом на задние колеса. Этот экзерсис даже возили на московский фестиваль «Автоэкзотика», где ему было самое место. Сейчас несколько подобных экземпляров доживают свой век в качестве внутризаводского транспорта. А универсал эволюционировал в пикап СеАЗ-1313 и был показан на MIMS’2005. Эта симпатичная серая машинка имела удлиненную базу (на 230 мм) и грузоподъемность 250 кг, что довольно неплохо, учитывая слабосильность ВАЗовского двухцилиндрового моторчика. Еще один пикап, зеленого цвета со стандартной базой, позиционировали как автомобиль для активного отдыха. Что и в названии отразилось – «Ока-Романтик». С этих двух машин и можно начинать официальное летосчисление «коммерческой» программы СеАЗа, которая во всей полноте явила себя на апрельской выставке «КомТранс-2007».
А универсал эволюционировал в пикап СеАЗ-1313 и был показан на MIMS’2005. Эта симпатичная серая машинка имела удлиненную базу (на 230 мм) и грузоподъемность 250 кг, что довольно неплохо, учитывая слабосильность ВАЗовского двухцилиндрового моторчика. Еще один пикап, зеленого цвета со стандартной базой, позиционировали как автомобиль для активного отдыха. Что и в названии отразилось – «Ока-Романтик». С этих двух машин и можно начинать официальное летосчисление «коммерческой» программы СеАЗа, которая во всей полноте явила себя на апрельской выставке «КомТранс-2007».
Китайские силы
Новую жизнь этому направлению, безусловно, дал китайский трехцилиндровый мотор TJ 3776 QE корпорации FAW (точнее – Tian Jin FAW Import and Export Corporation) мощностью 53 л.с., с начала этого года устанавливаемый в качестве базового. С ним «Ока» получила совершенно новые потребительские качества, которые и позволили всерьез задуматься о выпуске грузовых версий микролитражки.
| Предтеча всей грузовой программы СеАЗа – внутризаводской развозной пикап из некондиции |
Таких версий получилось четыре, условно их называют «универсал», «пикап», «фургон» и «грузовик».
Четыре модификации
«Ока-11116-0000011-50» – это пикап без тента на базе стандартного кузова «Оки» грузоподъемностью около 300 кг. У хэтчбека отрезается задняя часть крыши вкупе с задними стойками, а за центральными стойками вваривается глухая перегородка. На верхних торцах бортов сделаны элегантные «поручни», за которые можно крепить накидку, скрывающую груз или стропы грузофиксатора. Изменений в подвеску не вносили, поэтому грузовая платформа сильно поджата опорами амортизаторов и колесными арками. Зато сохранили низкую погрузочную высоту, опущенную до верхней кромки заднего бампера. Металлический задний борт откидывается на 180° и не мешает погрузке. Он фиксируется на подпружиненных верхних защелках. На пол кузова положена быстросъемная ламинированная фанера, а под ней, в нижней точке пола, специально удалили резиновые заглушки в сливных отверстиях.
| Мотор будут закрывать «европейской» крышкой |
Внешне машина получилась элегантной, динамичной. Да что там – просто красивой. Вот только практичность ее вызывает некоторый скепсис. Все-таки в наших условиях незащищенная грузовая платформа допустима только у мусоровоза. А для этого «Ока» никак не подходит – кузов не самосвальный, да и объем маловат. Так что название «Романтик» для этой «Оки» вполне подходит: она и смотрится игриво, и едет задорно. Но «пляжная» ипостась подразумевает люк во всю крышу, кондиционер, громкую музыку и даже полный привод. Всего этого у «Оки» с романтичным индексом 11116-0000011-50 нет и не предвидится. Так что будем считать ее транспортом для романтиков средней полосы России, где нет песчаных пляжей и вообще не очень жарко.
Универсал 11116-0000010-52, по сути, то же, что пикап, только с жесткой крышей без остекления. За задними сиденьями здесь та же глухая перегородка, а задняя дверь осталась подъемной, на двух газовых упорах (как и в хэтчбеке). Оконные проемы заделаны не очень аккуратно – по периметрам бывших окон остался рельеф. Конечно, вварить изнутри заглушки проще и технологичнее, чем сглаживать панели снаружи, но… Выглядит это на скорую руку.
Универсал, разумеется, получился тяжелее пикапа, поэтому груза он может взять меньше (официальных данных нет, но навскидку – на 70–80 кг). То есть он подходит для перевозки грузов небольшого объема и массы (до 0,8 «кубов») и идеально вписывается в специфику работы торговых представителей крупных компаний.
| Компоновка двигателя неидеальна, но агрегаты доступны для ремонта |
У них доставка осуществляется специализированным транспортом, а агенты возят с собой только образцы – шоколадки, жвачку, сигареты. .. Больше всего подходит эта машина для тех, кто возит мелкие партии тяжелых стройматериалов: глухая перегородка за сиденьями страхует от любых неожиданностей.
.. Больше всего подходит эта машина для тех, кто возит мелкие партии тяжелых стройматериалов: глухая перегородка за сиденьями страхует от любых неожиданностей.
Фургон 11116-0000010-50 (интересно, в чем практический смысл таких сложных цифровых комбинаций?) – это пикап с пластиковой надстройкой и подъемной дверью. Надстройка для увеличения внутреннего объема слегка выходит за границы «оковского» кузова. Невысокий металлический задний борт остался откидывающимся, поэтому тяжелые грузы высоко поднимать не придется. Подъемная дверь фиксируется по центру, а поднимается на тех же газовых упорах. Но открыть ее одной рукой не получится, так как нижняя кромка (за которую, собственно, и следует дверь поднимать) находится на приличном расстоянии от кнопки замка. Внутри кузов выглядит очень просторным (реально – 1,2 м 3), дополнительная полочка сделана над кабиной (снаружи ее прикрывает спойлер, уменьшающий Сх).
Занятно, что официальные источники дают те же показатели снаряженной и полной масс, что и для пикапа. Думается, это ошибка. Реальная грузоподъемность с одним водителем у фургона должна составлять примерно 330 кг. Подвеска-то здесь стандартная, пружинная.
Думается, это ошибка. Реальная грузоподъемность с одним водителем у фургона должна составлять примерно 330 кг. Подвеска-то здесь стандартная, пружинная.
| «Восьмерочный» вакуумник |
Грузовик 11116-60 – самая оригинальная машина «грузового» ряда СеАЗа. Это рамный автомобиль с кабиной (вернее, полукабиной) от «Оки». Кузов хэтчбека отрезается здесь по всему периметру, и к лонжеронам кабины приваривается оригинальная рама. Оригинальность в том, что делают ее не из трубчатого каркаса, а набирают из профилей, сваренных из обычной листовой стали и усиленными перегородками. Получается легкая и очень прочная конструкция с хорошей сопротивляемостью изгибу. Нам удалось заснять процесс рождения такого грузовика: в цехе на стапеле очень удачно стоял почти полностью сваренный кузов с подрамником. Это всего второй экземпляр этой модели; первый готовился для сертификационных испытаний и выставок.
У грузовика 11116-60 увеличенная на 47 см база и полностью оригинальная задняя подвеска – на длинных полуэллиптических рессорах. Соответственно кузов приподнялся над землей, но платформа теперь совершенно плоская, с аккуратными деревянными полозьями для легкого скольжения груза. Кузов объемом 2,3 м 3 имеет каркас для тента и сам тент с фиксацией петельками. Задний борт фиксируется настоящими грузовыми запорами (только маленькими) и откидывается на нижних петлях на те же 180°. Задний мост – балка прямоугольного сечения. Срез выхлопной системы расположился над левой рессорой, между ее задним креплением и амортизатором. Под рамой – мощный фаркоп, так что грузовик вполне подходит для буксировки не очень тяжелого прицепа. Оригинальные задние фонари сделала фирма «Евростандарт» из города Вязники. В задней стенке кабины здесь имеется окно, соответственно при снятии тента улучшается обзорность.
Под фанерным фальшполом пикапа – рифленый пол с углублением. В некоторых впадинах застаивается вода… В некоторых впадинах застаивается вода… |
Общее для всех
Как уже говорилось, единым для всех модификаций «Оки» стал китайский мотор с китайской же коробкой передач. Он смонтирован на оригинальном подрамнике, однако опоры остались старыми. Энергоемкость их явно недостаточна и даже на холостом ходу вибрации с избытком передаются на руль и рычаг переключения. Мотор свободно расположился в отсеке, так как «запаска» диаметром 13” туда не помещалась. Запасное колесо перекочевало за сиденье пассажира (единственного в двухместной машине), ограничив таким образом его ход. Места в кабине получилось достаточно даже для очень рослых людей.
Вернемся к двигателю. Он «заточен» под нормы Euro 2, поэтому оснащен каталитическим нейтрализатором и топливным абсорбером. Последний, равно как и топливный фильтр высокого давления, расположился на месте запасного колеса. Вообще компоновка вышла удобной с точки зрения доступности элементов, которым необходимо регулярное внимание, – расширительных бачков, свечей, фильтров. Но смущает некоторый хаос в магистралях, проложенных, как кажется, по кратчайшему пути и потому не всегда надежно закрепленных. С проводкой еще хуже: в некоторых местах разноцветное великолепие проводки ничем не защищено, даже изолентой жгут не обмотали, не говоря уже о пластиковых гофрированных трубках. Думается, что несколько недель эксплуатации превратят эту красоту в бесцветный комок грязи, с которым разобраться будет непросто.
Но смущает некоторый хаос в магистралях, проложенных, как кажется, по кратчайшему пути и потому не всегда надежно закрепленных. С проводкой еще хуже: в некоторых местах разноцветное великолепие проводки ничем не защищено, даже изолентой жгут не обмотали, не говоря уже о пластиковых гофрированных трубках. Думается, что несколько недель эксплуатации превратят эту красоту в бесцветный комок грязи, с которым разобраться будет непросто.
| Очень крутой подъем – процентов 45! – «Ока», побуксовав, осилила |
И еще один предмет – не столько смущения, сколько умиления: на внутренней стороне крышки капота наклеены фрагменты пенополиэтилена. Такая неграмотность в вопросах технической акустики простительна механикам с СеАЗа, но непростительна их подрядчикам по разработке схемы звукоизоляции – ивановской компании «Стандартпласт». Пенополиэтилен имеет закрытые поры, не продувается, и звуковая волна проходит сквозь него практически без потерь. По крайней мере, в спектре частот, который излучает силовой агрегат. Так что его применение в капотном пространстве можно назвать декоративно-теплосберегающим. К звукоизоляции, за которую потребитель, кстати, платит деньги, это отношения не имеет.
По крайней мере, в спектре частот, который излучает силовой агрегат. Так что его применение в капотном пространстве можно назвать декоративно-теплосберегающим. К звукоизоляции, за которую потребитель, кстати, платит деньги, это отношения не имеет.
Увеличившаяся масса машины потребовала ревизии тормозной системы. Первое, что приходит в голову всем и сразу: раз уж поставили большие колеса, то почему бы не поставить тормозные диски большего диаметра, от «Самары» например? На СеАЗе об этом тоже подумали. Подумали – и отказались. Точнее, отказались, когда посчитали, в какую сумму обойдется подготовка производства. Казалось бы просто заменить деталь. Но ее замена тянет за собой установку новых стоек, двух рычагов и поворотного кулака. На СеАЗе решили пойти по компромиссному пути: оставив диски и барабаны прежними, увеличить производительность системы путем установки «восьмерочных» вакуумного усилителя и главного тормозного цилиндра. В такой комплектации сделано несколько машин, и скоро это станет стандартным оборудованием всех модификаций – и легковых, и грузовых.
| Шины подобраны очень удачно |
Обновления в салоне затронули сиденья и приборную панель. Новые кресла значительно удобнее прежних бесформенных «оковских» сидений, спинки и подушки которых больше напоминали черенки огромных дворницких лопат, даром что обшитых кожзамом. Нынешние кресла обиты хорошей тканью, имеют удобную конфигурацию и неплохо распределяют нагрузки. В них уже не устанешь в первые полчаса. Спинка регулируется классической вращающейся рукояткой, а фиксатор полозьев управляется не менее классическим рычажком под подушкой. Немного не хватает разве что поясничного подпора. Но все портит невнимание «сеазовцев» к качеству изготовления: швы порой грубы, из-под обшивки местами просматривается мягкий наполнитель, а механизм откидывания спинки работает через пеньколоду. Пальцы можно сломать, дергая за неудобный рычаг защелки! Оказавшись в одиночку на заднем сиденье (речь конечно же об обычной четырехместной «Оке»), нечего и пытаться выйти из машины. Для грузовиков это не очень актуально, но осадок остался.
Для грузовиков это не очень актуально, но осадок остался.
Эксперименты с панелями приборов начались на СеАЗе давно, но ничего лучше первоначального ВАЗовского варианта сделать не получалось. Концептуальная панель на базе приборов от ВАЗ-2106 вышла столь уродливой, что надолго стала объектом насмешек. Недавно договорились с двумя фирмами из Тольятти и Набережных Челнов («СКС-Тольятти» и «АвтоСтильСервис» соответственно), сейчас они поставляют два новых варианта панелей на базе комбинации приборов «десятки», и оба идут на конвейер. Панель СКС немного дороже, но сделана лучше и постепенно вытесняет челнинскую. Однако претензии по качеству изготовления и конструкции есть к обеим.
| Умеренные крены и минимальные уводы шин дают «Оке» надежную управляемость |
Панель от «АвтоСтильСервис» «радует» обилием неприкрытых саморезов – они держат рамку комбинации приборов и накладки на центральную консоль. Пластик довольно хлипкий, и если верхняя часть с натяжкой может именоваться «мягкой», то консоль и крышка бардачка сделаны просто безобразно, с неровными стыками, облоем и заусенцами на краях. Бардачок выглядит совсем неприглядно – крышка болтается на хлипенькой оси, а «пятёрочный» замок защелкивается только после крепкого удара. Не лучше и накладка на тоннель, под рычаг «ручника».
Пластик довольно хлипкий, и если верхняя часть с натяжкой может именоваться «мягкой», то консоль и крышка бардачка сделаны просто безобразно, с неровными стыками, облоем и заусенцами на краях. Бардачок выглядит совсем неприглядно – крышка болтается на хлипенькой оси, а «пятёрочный» замок защелкивается только после крепкого удара. Не лучше и накладка на тоннель, под рычаг «ручника».
Кроме основного воздуховода, отвечающего за обдув ветрового стекла, челнинская панель имеет два маленьких сопла по бокам, воздушный поток из которых фиксированно направлен в область зеркал заднего вида. Это, конечно, хорошо, но исчезли дефлекторы индивидуального обдува, так что водитель с пассажиром могут направить воздух только себе в ноги, через отдельную «жигулевскую» заслонку.
Дизайн этой панели сделан явно по мотивам новой «десятки», но в «пухлости» создатели немного перегнули планку. Они не учли, что «Ока» – машина маленькая, здесь каждый сантиметр на вес золота. Право же, не стоило центральную консоль выдавать так далеко в салон, надо бы поскромнее.
Панель из Тольятти выглядит более гармонично, в ней нет торчащих головок саморезов и дрожащих на сквозняке тонких пластиковых накладок. Перчаточный ящик крышки не имеет вовсе, и это, как ни странно, довольно удобно. Качество изготовления лучше, но тоже не без огрехов. Например, рамка приборов стыкуется с консолью с зазором 5–6 мм! «Борода», хоть и менее массивная, тоже выпирает в салон, и здесь это кажется более значительным просчетом, так как углы у нее острые и передний пассажир все время ударяетя об угол левой коленкой.
| «Запаске» нашлось место за пассажирским сиденьем |
И ладно бы эта «борода» несла значительную функциональную нагрузку! Нет, в ней расположена пепельница, от которых все мировые автопроизводители постепенно отказываются, внося, таким образом, посильный вклад в борьбу с курением.
Прямых регулируемых дефлекторов нет и здесь, но боковые сопла воздуховодов прикрыты изящными сеточками, имитирующими грили высокочастотных динамиков. Вот только куда направляются воздушные потоки, не совсем понятно. Да и какие там потоки… На высшей скорости вентилятора поток воздуха их боковых отверстий ощущается ладонью едва-едва.
Вот только куда направляются воздушные потоки, не совсем понятно. Да и какие там потоки… На высшей скорости вентилятора поток воздуха их боковых отверстий ощущается ладонью едва-едва.
Из нововведений стоит упомянуть новые внутренние дверные ручки и релинги на крыше, которыми комплектуется большинство серийных машин. Практичность последних неочевидна, но с релингами «Ока» смотрится динамичнее – факт.
На дороге
К сожалению, в день нашего визита на СеАЗ длиннобазный рамный грузовик существовал в одном экземпляре. Его берегли как музейный раритет и нам отказали в удовольствии дорожного знакомства, но фургон и хэтчбек удалось узнать в деле.
Первое, на что вынужден обратить внимание всякий, не избалованный трехдверными автомобилями человек, – задняя стойка отнесена назад, поэтому за ремнем безопасности приходится тянуться. Но на некоторых американских машинах эту проблему решают введением небольшого промежуточного качающегося кронштейна, удерживающего наконечник лямки поблизости от водителя. Почему бы не сделать так на «Оке»?
Почему бы не сделать так на «Оке»?
| Фиксатор откидывающегося заднего борта |
Передачи включаются четко и понятно, выбор их не доставляет никаких хлопот – такое ощущение, что рычаг расположен прямо на картере коробки и нет никаких промежуточных тяг. Но дают о себе знать скромные габариты: рычаг смещен немного назад против привычного для большинства водителей уровня и требует некоторого привыкания. И нужно учесть, что ходы рычага между передачами здесь меньше, чем обычно, поэтому и расстояние «между передачами» меньше ожидаемого, работать рычагом нужно очень точно. Вначале промашки неизбежны и потому извинительны.
Еще большего привыкания потребует педаль сцепления. Информативность ее стремится в отрицательную область: уже кажется выбрал весь ход, уже «газуешь» с запасом, ожидая неизбежного рывка… а ничего не происходит. И лишь в самом конце хода, когда начинаешь проверять, а включена ли передача, машина неожиданно и резво трогается. Совсем непонятно, зачем педали сцепления сообщили такой большой свободный ход при таком маленьком рабочем и полном отсутствии информирующего усилия. Есть над чем поработать.
Совсем непонятно, зачем педали сцепления сообщили такой большой свободный ход при таком маленьком рабочем и полном отсутствии информирующего усилия. Есть над чем поработать.
| Оригинальные задние фонари из Вязников |
Фургончик стартует активно, шумно. В принципе, адаптировавшись, трогаться можно на минимальных оборотах и небрежно бросая сцепление – «моментный» мотор вывезет. Но если тянет немного похулиганить, то «Ока» всеми четырьмя колесами «за!». Ничего, что за спиной пластиковая будка с большими площадью фронтальной проекции и парусностью. За темпом набора скорости и послушным повизгиванием шин в поворотах об этом не задумываешься. Моторчик прощает даже грубые ошибки в выборе передач (момента хватает), а при точном их выборе разгоняет машинку с непривычно высокой скоростью. Что ж, соотношение массы и мощности здесь примерно такое же, как в однотонном седане С-класса со стосильным двигателем, т. е. такое, которое не вынуждает раскручивать коленвал до обжигающих звуков детонации.
е. такое, которое не вынуждает раскручивать коленвал до обжигающих звуков детонации.
Китайский двигатель малочувствителен к загрузке. Он довольно рано, едва ли не с холостых оборотов выходит на пик крутящего момента и сохраняет тягу в широком диапазоне оборотов. Подобный характер был бы понятен у американского восьмицилиндрового гиганта объемом четыре-пять литров, с длиннющим ходом поршня и невысокой степенью сжатия. Но откуда такая ровная характеристика в маленьком «китайце»? Воистину, не все в этом мире объяснимо.
| Панель из Набережных Челнов: спорный дизайн, неприкрытые головки саморезов, хлипкий пластик, неудобный «бардачок» |
Теоретически центр тяжести должен был подняться. Но на управляемости это никак не отразилось. Крены при прохождении поворотов минимальны, подвеска сопротивляется до последнего. А когда дошел до последнего… Заводской испытатель с удовольствием показал нам, что «Ока» остается управляемой даже при отрыве колес одного из бортов, и вернуть ей строгое горизонтальное положение совсем несложно. Явной склонности к опрокидыванию у фургона никто из нас не почувствовал. Скажем больше: то, что основная масса машины находится в пределах колесной базы, обеспечивает ей отменную устойчивость далеко за гранью паспортных характеристик. Например, при максимальном заявленном подъеме 30% упомянутый испытатель легко забирался на горку крутизной под 45%! С пробуксовкой, ревом двигателя, облаками пыли из-под колес, но забирался. Машине всегда хватало сцепного веса!
Явной склонности к опрокидыванию у фургона никто из нас не почувствовал. Скажем больше: то, что основная масса машины находится в пределах колесной базы, обеспечивает ей отменную устойчивость далеко за гранью паспортных характеристик. Например, при максимальном заявленном подъеме 30% упомянутый испытатель легко забирался на горку крутизной под 45%! С пробуксовкой, ревом двигателя, облаками пыли из-под колес, но забирался. Машине всегда хватало сцепного веса!
Но надо признать, что рулевое управление у машины могло быть и построже. Само шасси безупречно выполняет команды, но пока эти команды до него дойдут… Свободный ход рулевого колеса в «околонулевой» зоне пугающе велик, на его выбор тратится драгоценное время маневра. Потом, когда реечный механизм все же принялся за дело, машина управляется надежно и предсказуемо, например, в затяжных поворотах с доворотом или при активном рулении. Главное не ставить руль в «нуль»!
То есть при прямолинейном движении рыскания, обусловленные отсутствием информации о положении колес, обеспечены. Обеспечен и широкий динамический коридор, с которым можно мириться примерно до 100 км/ч. Потом удержание траектории требует изрядной концентрации. И еще вибрации! Бог ведает, какие особенности конструкции заставляют руль заходиться в пароксизме дрожи – может быть, неподходящие опоры агрегата (но тогда биения больше зависели бы от оборотов, нежели от скорости), может, банальная разбалансировка колес… Но оба экземпляра, выданные нам на тест, после 100 км/ч превращались в зубодробительный станок. Некомфортно.
Обеспечен и широкий динамический коридор, с которым можно мириться примерно до 100 км/ч. Потом удержание траектории требует изрядной концентрации. И еще вибрации! Бог ведает, какие особенности конструкции заставляют руль заходиться в пароксизме дрожи – может быть, неподходящие опоры агрегата (но тогда биения больше зависели бы от оборотов, нежели от скорости), может, банальная разбалансировка колес… Но оба экземпляра, выданные нам на тест, после 100 км/ч превращались в зубодробительный станок. Некомфортно.
Обзорность через узенькие штатные зеркала оказалась на удивление неплохой. Несмотря на скромные габариты зеркала за счет удачной вытянутой формы отображают необходимое для маневров пространство по бокам автомобиля, оказывая при этом минимальное аэродинамическое сопротивление. А кому аэродинамика не важна, кто не гоняет на грузовиках ради кайфа и адреналина, можно посоветовать зеркала от «Жигулей» (российские или турецкие). У них площадь существенно больше и такие зеркала будут ставить на рамный грузовик.
Тормоза сюрпризом не стали. Педаль заметно сопротивляется потяжелевшей машине, но сверхусилий не требует, колом не встает и позволяет с достаточной точностью дозировать замедление. Траекторные рыскания при этом не выходят за рамки разумного. На обычной «Оке» уже стоял усилитель большей производительности (тот самый, «восьмёрочный»), и с ним торможение происходит более плавно.
Конечно, есть разница в ощущениях, но она не катастрофична, ее больше хрупкие женщины отметят. Но будут ли хрупкие женщины массово скупать себе фургоны и грузовики «Ока»? Вряд ли. Да и штатным «оковским» тормозам срок уже отмерен.
Отдельно хочется похвалить выбор шин. Вроде бы невзрачные покрышки «Росава ВС-11» размерности 155/70 R13 отлично цепляются за асфальт и грунт, имеют незначительные боковые уводы, надежны при торможении – не блокируются почем зря. Это всяко лучше КАМЫ, которой «Оку» комплектовали раньше. Соотношение ширины и профиля подобрано, кажется, идеально – и не трясет, и в поворотах не страшно, и сопротивление качению небольшое. И еще одно замечание: признаться, мы ожидали, что при вертикальной раскачке большие колеса будут задевать за арки. Так вот, ничего подобного. Мы, правда, не загружали машину балластом (но вдвоем, а в хэтчбеке вчетвером), но никакого намека на то, что колесам тесно в арках, мы не получили. Тем, кто собирается эксплуатировать «Оку» с перегрузом, можно посоветовать поставить шины с меньшей высотой профиля, но пошире.
Соотношение ширины и профиля подобрано, кажется, идеально – и не трясет, и в поворотах не страшно, и сопротивление качению небольшое. И еще одно замечание: признаться, мы ожидали, что при вертикальной раскачке большие колеса будут задевать за арки. Так вот, ничего подобного. Мы, правда, не загружали машину балластом (но вдвоем, а в хэтчбеке вчетвером), но никакого намека на то, что колесам тесно в арках, мы не получили. Тем, кто собирается эксплуатировать «Оку» с перегрузом, можно посоветовать поставить шины с меньшей высотой профиля, но пошире.
Если суммировать ездовые впечатления, то получится вот что: машина разболтана, не без огрехов в настройках и качестве изготовления, но с огромным потенциалом. А выбор мотора – большая удача СеАЗа. Он подходит для «Оки» на 100% по всем параметрам.
Что же дальше?
| Панель из Тольятти: дизайн интереснее, качество выше, но тоже не без проблем |
А дальше, буквально через полгода, наступит первое января. И это будет дата введения в России обязательного соответствия всех выпускаемых на ее территории автомобилей экологическим нормам Еuro 3. СеАЗ с этой новостью оказался в непростом положении: только-только начали ставить Еuro 2 – и опять нужно вкладывать средства в модернизацию. Но, к счастью, мотор Tian Jin её допускает, причем относительно небольшими затратами. Сейчас заводчане рассматривают два варианта выхода из положения. Первый заключается в том, чтобы получать от китайцев уже адаптированный под Еuro 3 мотор. Конечно, он будет дороже нынешнего, но удорожание вообще неизбежно, с этим придется смириться. А второй вариант связан с предложением некой московской фирмы, которая берется сделать новый контроллер (он будет немного дешевле китайского!), ПО и поставить два дополнительных датчика – фаз и кислорода (второй лямбда-зонд, после катализатора). После такой доработки двигатель будет оснащен программой диагностики, работающей по международному протоколу OBD-2, а это значит, чт
И это будет дата введения в России обязательного соответствия всех выпускаемых на ее территории автомобилей экологическим нормам Еuro 3. СеАЗ с этой новостью оказался в непростом положении: только-только начали ставить Еuro 2 – и опять нужно вкладывать средства в модернизацию. Но, к счастью, мотор Tian Jin её допускает, причем относительно небольшими затратами. Сейчас заводчане рассматривают два варианта выхода из положения. Первый заключается в том, чтобы получать от китайцев уже адаптированный под Еuro 3 мотор. Конечно, он будет дороже нынешнего, но удорожание вообще неизбежно, с этим придется смириться. А второй вариант связан с предложением некой московской фирмы, которая берется сделать новый контроллер (он будет немного дешевле китайского!), ПО и поставить два дополнительных датчика – фаз и кислорода (второй лямбда-зонд, после катализатора). После такой доработки двигатель будет оснащен программой диагностики, работающей по международному протоколу OBD-2, а это значит, чт
какими были первые поезда в метро / Новости города / Сайт Москвы
История развития столичного метрополитена — это хроника промышленных рекордов, технологического прогресса и непрерывной адаптации к новым вызовам. В день открытия метро в Москве пассажирам были доступны 13 станций. А сейчас столичная подземка — одна из крупнейших в мире. Это 275 станций с учетом станций Московского центрального кольца. За 85 лет на линии метро выходили поезда более десяти различных типов и модификаций.
В день открытия метро в Москве пассажирам были доступны 13 станций. А сейчас столичная подземка — одна из крупнейших в мире. Это 275 станций с учетом станций Московского центрального кольца. За 85 лет на линии метро выходили поезда более десяти различных типов и модификаций.
За последние десять лет парк вагонов рос и обновлялся как никогда раньше. Четыре типа поездов запускали в 2010–2019 годах. Это последние модификации номерных поездов, а также поезда «Русич», «Ока» и «Москва». Столичное метро вошло в десятилетие с 12,6 процента новых поездов, а к 2020-му парк был обновлен на 57 процентов за счет покупки 2997 вагонов.
В конце марта на Некрасовской и Большой кольцевой линиях запустили еще 14 поездов нового поколения «Москва». Составы начали курсировать после открытия 27 марта станций «Юго-Восточная», «Окская», «Стахановская», «Нижегородская», «Авиамоторная» и «Лефортово». В инновационных поездах созданы комфортные условия для пассажиров: в них широкие дверные проемы, есть сквозной проход через все вагоны и увеличенные электронные табло с экранами высокого разрешения, а для обеззараживания воздуха установлены ультрафиолетовые лампы.
А mos.ru предлагает проследить, какой путь прошли поезда метро — от самых первых до инновационных поездов «Москва».
А и БПервые поезда Московского метрополитена собрали в 1934 году на Мытищинском машиностроительном заводе специально к открытию подземки. К началу регулярного движения город получил 58 вагонов серии А. В каждом из них были мягкие диваны с кожаной обивкой — всего 52 сидячих места. В общей сложности вагон вмещал около 260 человек.
Кстати, первые вагоны типов А и Б были песочно-желтыми — светлый верх и темно-красный, почти кирпичный низ. В процессе эксплуатации они были перекрашены в более привычные цвета: зеленый и синий.
До января 1936 года в метро работали четырехвагонные составы, ходившие с пятиминутным интервалом. Затем пустили шестивагонные поезда. Производство вагонов серии А прекратилось в 1937 году. В метро они ходили в общей сложности около 40 лет. Затем их изъяли из эксплуатации.
Сегодня в электродепо «Измайлово» есть состав из вагонов серии А. В 2015 году, к 80-летию столичного метро, их полностью восстановили.
В 2015 году, к 80-летию столичного метро, их полностью восстановили.
Первые вагоны серии Б изготовили в 1937 году для линий, которые открывали в рамках второй очереди строительства метрополитена, — Покровского радиуса со станциями «Площадь Революции» и «Курская», а также Горьковского радиуса (участок нынешней Замоскворецкой линии) со станциями «Площадь Свердлова» («Театральная»), «Маяковская», «Белорусская», «Динамо», «Аэропорт» и «Сокол».
По сути, это были усовершенствованные вагоны серии А. Габаритные размеры, колесные пары, тяговые электродвигатели и мотор-компрессоры секций серий А и Б были одинаковыми, а остальное оборудование имело лишь небольшие различия.
При этом в вагонах типа Б была улучшена система вентиляции, что обеспечивало более равномерный обмен воздуха по всему салону. Вагоны этой серии эксплуатировали до 1976 года.
Г — ГорьковскийЭти вагоны получили буквенное обозначение Г, потому что предназначались для эксплуатации на Горьковской линии. Первые шесть вагонов произвели в 1940 году, однако из-за войны серийное производство удалось наладить только спустя семь лет.
Первые шесть вагонов произвели в 1940 году, однако из-за войны серийное производство удалось наладить только спустя семь лет.
Вагоны серии Г были на восемь тонн легче по сравнению с вагонами типа А и Б. Главное их отличие — все они стали моторными и были оснащены новым электрооборудованием, а также электрическим торможением.
На момент разработки вагон серии Г был одним из самых современных вагонов метро в мире как по технико-эксплуатационным и динамико-скоростным показателям, так и по надежности работы.
Новые станции метро и современные вагоны: достижения транспортной системы и планы на 2020 годМосквичи поддержали программу строительства метро
В — военныйК концу Великой Отечественной войны, в 1945 году, в столичном метро насчитывалось всего 278 вагонов. В условиях растущего пассажиропотока и в связи с открытием новых станций в годы войны стала ощущаться нехватка подвижного состава.
Поэтому параллельно с организацией производства вагонов серии Г в 1945–1946 годах модернизировали немецкие электровагоны типов С1 и С2. Их доставили в Москву в 1945-м в количестве 120 штук из Берлинского метрополитена. Эти вагоны получили обозначение В — военный.
Их доставили в Москву в 1945-м в количестве 120 штук из Берлинского метрополитена. Эти вагоны получили обозначение В — военный.
До модернизации двери в них автоматически только закрывались, а открывали их изнутри пассажиры. Сиденья в вагонах были обиты бордовым плюшем. Его сняли и сделали шнуры, которые затем использовали для ограждения эскалаторов во время ремонта.
Для пассажиров московского метро обивку сидений сделали кожаной. Вместо деревянной обшивки использовали линкруст — плотную бумагу с вытесненными на ней узорами.
Сперва иностранные вагоны курсировали на линии «Сокольники» — «Парк культуры». Затем часть из них перебросили на Калининскую (ныне — Филевскую) линию, предварительно утеплив пассажирские салоны (между станциями «Киевская» и «Фили» пути находятся на поверхности).
В начале 1960-х все вагоны серии В списали. Часть переделали в электровозы и путеизмерители для хозяйственных нужд.
Д и Е — по алфавитуОпытную партию вагонов серии Д выпустили в 1949 году под обозначением М5 (метровагон пятого поколения). После усовершенствования эти вагоны получили обозначение УМ5. Серийно их производили с 1955 по 1963 год.
После усовершенствования эти вагоны получили обозначение УМ5. Серийно их производили с 1955 по 1963 год.
Название Д таким вагонам присвоили просто потому, что предшествовавшие серии — А, Б, В и Г — случайным образом повторили алфавитный порядок. Пассажирская эксплуатация новых вагонов началась в 1956 году. Они курсировали в метро до 1995-го.
Визуально они мало отличались от вагона серии Г: разве что уменьшилось количество сидячих мест, это позволило сделать карманы возле дверей для стоящих пассажиров. В то же время масса вагонов серии Д уменьшилась на семь тонн, за счет чего снизилось потребление электроэнергии. Также были различия в ходовой части, электрическом и механическом оборудовании.
Вагоны серии Д курсировали на Кировско-Фрунзенской (сейчас — Сокольническая), Арбатско-Покровской, Филевской и Калужско-Рижской линиях.
Вагоны серии Д выпускали до 1963 года, после чего Мытищинский машиностроительный завод стал производить вагоны типа Е. Общий парк подвижного состава в Московском метрополитене к тому времени вырос более чем в 3,5 раза по сравнению с 1945 годом и достиг 1078 вагонов.
Опытный состав серии Е выпустили в 1959–1960 годах — их было всего семь. Серийное производство велось с 1963 по 1969 год. Кстати, прокатиться в вагоне этой серии можно было вплоть до 2008-го.
У новых вагонов появились полосы гофрированного металла на корпусе: обшивка стала прочнее, аэродинамика улучшилась. Впервые в вагонах расширили дверные проемы, что позволило ускорить посадку и высадку пассажиров. Из-за этого вагон потерял свою симметричность: дверные проемы сместились относительно центра кузова. Впервые использовали алюминиевые сплавы, из которых готовили двери и другие элементы кузова.
Вагоны серии Е значительно отличались от предшественников не только внешне, но и динамическими и ходовыми свойствами. Так, конструкционная скорость выросла до 90 километров в час. Это обеспечило более высокую пропускную способность линий метро.
Эти вагоны курсировали на Сокольнической, Замоскворецкой, Арбатско-Покровской, Филевской, Калужско-Рижской и Таганско-Краснопресненской линиях. Кроме московской подземки, вагоны серии Е ездили в Ленинграде, Киеве, Тбилиси и Баку.
Кроме московской подземки, вагоны серии Е ездили в Ленинграде, Киеве, Тбилиси и Баку.
Появившиеся в 1960-е в Ленинграде станции новой конструкции не позволяли эксплуатировать вагоны серии Е. Появилась задача разработать новый вагон с аналогичными параметрами. В результате дверные проемы в вагонах снова стали симметричными за счет изменения посадочных мест, а также ликвидации сидений возле кабины машиниста. В отделке новых вагонов вместо линкруста использовали новый материал — слоистый пластик.
Вагоны серий Ем и Еж были полностью совместимы с вагонами Е. Их часто можно было встретить в одном составе.
В московском метро прошедшие модернизацию вагоны Еж-3 до сих пор эксплуатируются на Таганско-Краснопресненской линии. По состоянию на начало апреля 2020 года на линии курсируют всего два таких состава. Еще четыре — в резерве. Скоро их все заменят поезда «Москва».
Быстрые, удобные и современные: метро получит еще около 700 вагонов для поездов «Москва»Сергей Собянин: В течение года планируем полностью обновить подвижной состав МЦД
НомерныеСамая массовая серия вагонов не только в истории столичного метро, но и в мире — 81-717/714. Их часто называют номерными. Их и сегодня можно встретить в 18 метрополитенах по всему земному шару.
Их часто называют номерными. Их и сегодня можно встретить в 18 метрополитенах по всему земному шару.
Первые номерные вагоны произвели в 1976 году, а в 1978-м их выпустили на линии метро. Усовершенствованная электрическая схема позволила снизить расход энергии на пять-шесть процентов. Изменились экстерьер и интерьер вагонов. Кстати, в салонах номерных вагонов впервые появилось люминесцентное освещение. А за счет того, что в промежуточных вагонах перестали размещать кабину машиниста, увеличилась их вместимость. В общей сложности провозная способность поезда выросла на 10 процентов.
Номерные вагоны выпускались с устройствами автоматического регулирования скорости и экстренной связи пассажиров с машинистом.
Сегодня в эксплуатируемом парке Московского метрополитена насчитывается более 2,3 тысячи таких вагонов. Они работали на всех линиях за исключением Арбатско-Покровской и Некрасовской. Сегодня они перевозят пассажиров Сокольнической, Замоскворецкой, Калужско-Рижской и Люблинско-Дмитровской линий.
Первый состав из вагонов типа 81-720/81-721 («Яуза») собрали в 1990–1991 годах. Они курсировали с 1998 по 2019 год на Люблинско-Дмитровской и Каховской линиях.
Главное их отличие от всех предыдущих моделей — новая система управления тяговым приводом. Она позволяет поезду плавно набирать и сбрасывать скорость.
На второй модификации «Яузы» впервые в отечественном серийном метровагоностроении установили асинхронный тяговый двигатель. Такой электропривод используется на всех более современных поездах метро.
Максимальная конструктивная скорость выросла до 100 километров в час. Вместимость вагонов увеличилась на 30–40 человек за счет уменьшения числа сидячих мест и образования вместительных торцевых площадок. Впервые были сделаны сиденья, повторяющие изгиб спины человека.
Также вагоны «Яуза» оборудовали микропроцессорной системой автоматического управления и технической диагностики, электропневматической системой управления тормозами, новыми системами пожаротушения и пожарной сигнализации, экстренной связи и радиооповещения пассажиров. На случай эвакуации пассажиров предусмотрели аварийный трап.
На случай эвакуации пассажиров предусмотрели аварийный трап.
Из салонов «Яузы» исчезли хорошо знакомые пассажирам форточки и воздуховоды: их заменила принудительная система вентиляции с малошумными агрегатами.
«Русич»Серийное производство «Русичей» (тип 81-740/81-741) велось с 2003 по 2013 год. В столичной подземке насчитывается 724 таких вагона: они курсируют на Арбатско-Покровской, Кольцевой и Бутовской линиях.
Стояла задача сделать подвижной состав для работы на наземных линиях. Разработка и постройка первого опытного состава из двух вагонов заняли рекордные для того времени полтора года. За технологическую основу были взяты узлы вагонов «Яуза», но сочлененную конструкцию кузова применили впервые. Такая конструкция позволяет проходить повороты без снижения скорости.
Дизайн салона кардинально переработали. В окнах установили двойные ударопрочные стеклопакеты с тонированными стеклами. Сделали антивандальные диваны с мягкими вставками, многослойный пол, а двустворчатые двери впервые стали прислонно-сдвижными, что позволило обеспечить их герметичное прилегание к корпусу вагона.
Эти меры позволили значительно снизить уровень шума в салоне при движении.
Еще одно новшество — в пассажирском салоне установили систему отопления, а затем и кондиционеры. Позже были разработаны аналогичные вагоны и для подземных линий с увеличенным количеством дверей.
«Ока»Поезда из вагонов серии 81-760/761 («Ока») выпускались с 2010 по 2016 год. Состав презентовали в день открытия станции «Марьина Роща» 19 июня 2010 года. Но массово серия «Ока» начала эксплуатироваться в подземке только в 2012-м.
Сейчас в столице насчитывается 1312 таких вагонов. Они ходят по Серпуховско-Тимирязевской, Калининской, Солнцевской и Большой кольцевой линиям.
В салонах установлены кондиционеры, в головных вагонах предусмотрено место для размещения одной инвалидной коляски или коляски для детей. Стены, потолок и пол облицованы легкомоющимися материалами, удовлетворяющими требованиям пожарной безопасности и санитарно-гигиеническим нормам.
Впервые именно в этой модели сделали сквозной проход между вагонами.
«Москва»В апреле 2017 года в метро начали курсировать отечественные поезда нового поколения — «Москва» (серии 81-765/766/767). Поезда «Москва» надежны, комфортны и более вместительны, чем их предшественники.
Поезда «Москва» сегодня занимают второе место по распространенности в столичной подземке — они составляют более 20 процентов, уступая лишь номерным. Чаще всего их можно встретить на платформах станций Таганско-Краснопресненской линии — здесь ходит около 80 таких составов, а также на Сокольнической, Филевской, Калужско-Рижской, Солнцевской, Некрасовской и Большой кольцевой линиях.
В поездах есть сквозной проход через все вагоны. Они удобны для маломобильных пассажиров: в них более широкие дверные проемы, а в головных вагонах предусмотрены специальные места для инвалидных колясок. Кроме того, в вагонах можно заряжать смартфоны и планшеты с помощью USB-портов, а сенсорные мониторы на стенах помогают найти нужные станции, проложить маршрут и рассчитать время в пути.
В прошлом году в метро появились поезда «Москва» новой модификации («Москва-2019») с системой электродинамического торможения. Она обеспечивает плавное снижение скорости поезда вплоть до его полной остановки.
В обновленных вагонах над дверями появились увеличенные электронные табло с экранами высокого разрешения. На них проще увидеть названия станций. В составах установлены новые поручни в форме шестиугольника. Они расположены на потолке вагона, рядом с дверями. Такие поручни не занимают много места и удобны для тех, кто стоит у дверей и готовится к выходу.
Самые дешевые автомобили на российском рынке. Что выбрать? :: Autonews
Самые дешевые автомобили на нашем рынке – это либо возрастные российские модели, либо новые китайские:ВАЗ-11116 “Ока” – $5170
ВАЗ-2105 “Жигули” – $5200
ВАЗ-2107 “Жигули” – $5650
BYD Flyer – $5990
Daewoo Matiz – $7690
Chery Sweet – $7400
Hafei Brio – $7640
ВАЗ-2113 “Самара-2” – $7850
“Ока” (ВАЗ-1111) впервые была выставлена в 1987 году на ВДНХ. Скромных размеров транспортное средство претендовало на роль народного автомобиля, и на тот период его кузов выглядел ладным и аккуратным, а интерьер салона был прост и дешев. Автомобиль полностью соответствовал своей задаче – обеспечить приемлемый комфорт при небольшой цене. В серию “Ока” пошла два года спустя с 650-кубовым двухцилиндровым мотором, развивавшим 30 л.с. Это был самый дешевый автомобиль еще в Советском Союзе.
Скромных размеров транспортное средство претендовало на роль народного автомобиля, и на тот период его кузов выглядел ладным и аккуратным, а интерьер салона был прост и дешев. Автомобиль полностью соответствовал своей задаче – обеспечить приемлемый комфорт при небольшой цене. В серию “Ока” пошла два года спустя с 650-кубовым двухцилиндровым мотором, развивавшим 30 л.с. Это был самый дешевый автомобиль еще в Советском Союзе.
Это и до сих пор самый дешевый автомобиль, но из массового продукта “Ока” превратилась в штучный товар. На фоне других конкурентов ее размеры, отделка, уровень безопасности просто не соответствуют даже минимальным требованиям покупателей.
После введения в России норм “Евро-2” на Заводе микролитражных автомобилей в Набережных Челнах решили дать отставку “Оке” со старым двигателем и прекратили ее выпуск. А на заводе в Серпухове старые моторы “Оки” заменили на китайские двигатели. Теперь у “Оки” трехцилиндровый силовой агрегат объемом 1 литр и мощностью 53 л. с. Самый дешевый автомобиль на нашем рынке стоит $5170 (137 400 руб).
с. Самый дешевый автомобиль на нашем рынке стоит $5170 (137 400 руб).
Чуть дороже, чем “Ока” с китайским мотором, еще один автомобиль тольяттинских конструкторов из далекого советского прошлого – ВАЗ-2105. Начало серийного производства – 26 лет назад. В прародителях у “пятерки” основа от полуитальянской “копейки” и переработанный на современный лад, для 80-го года, кузов FIAT 128. Удобств для водителя на “пятерке” немногим больше, чем на “Оке”. Но размеры автомобиля уже позволяют говорить о некоторой степени солидности и нормально разместить в салоне четырех человек. При этом за лишний метр железа кузова и дополнительный цилиндр мотора доплачивать нужно совсем немного – около $30. Цена на ВАЗ-2105 с инжекторным двигателем начинается от $5260. За кузов универсал (ВАЗ-2104) просят на 1000 долл. дороже.
ВАЗ-2107 отличается от “пятерки” только слегка благоустроенным салоном. Это люксовая и к середине 80-х самая дорогая модель АвтоВАЗа. На этом автомобиле ездили маленькие партийные чиновники.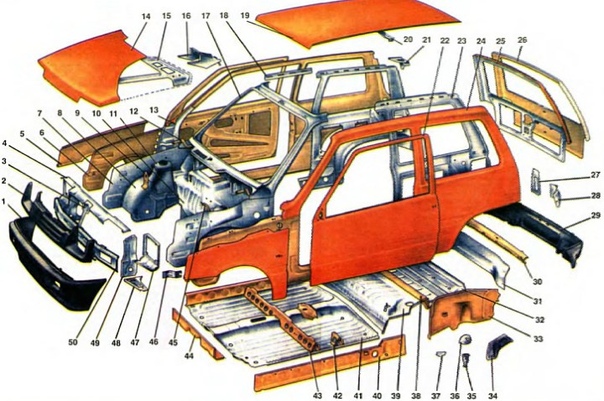 Большие бонзы предпочитали “Волгу”. Имидж солидного автомобиля, приобретенный “Волгой” в советские времена, сегодня вывел этот автомобиль вместе с его старой конструкцией за рамки самых дешевых моделей. А цены на ВАЗ-2107 с инжекторным двигателем 1,5 литра начинаются от 5650 долл.
Большие бонзы предпочитали “Волгу”. Имидж солидного автомобиля, приобретенный “Волгой” в советские времена, сегодня вывел этот автомобиль вместе с его старой конструкцией за рамки самых дешевых моделей. А цены на ВАЗ-2107 с инжекторным двигателем 1,5 литра начинаются от 5650 долл.
Китайская модель 2001 года BYD Flyer у нас на рынке стала самой дешевой из импортируемых автомобилей. Китайская конструкция явилась развитием некогда выпускавшейся лицензионной копии японского аналога Suzuki Alto из 90-х. Салон – уже и вовсе самостоятельная разработка, где используются вставки под дерево – попытка повторить некогда популярное почти у всех мировых производителей увлечение пластмассой под дерево.
Специально для российского рынка в конструкцию автомобиля были внесены изменения, максимально облегчающие эксплуатацию в холодное время года. Цена на иностранную модель всего на 750 долл. больше, чем цена на “Оку”. При этом разница моделей в возрасте, в используемых материалах и в дизайне разительна. У китайского авто четыре боковые двери, и в базе идет магнитола. Но BYD обладает худшими на нашем рынке характеристиками: самым слабым мотором в 40 л.с., самым медленным разгоном до 100 км/ч – 25 сек. и максимальной скоростью в 118 км/ч. Помнится, такая же цифра стояла в паспортной книжке дедушкиного “Запорожца” ЗАЗ-968М. BYD Flyer в минимальной комплектации стоит 5990 долл.
У китайского авто четыре боковые двери, и в базе идет магнитола. Но BYD обладает худшими на нашем рынке характеристиками: самым слабым мотором в 40 л.с., самым медленным разгоном до 100 км/ч – 25 сек. и максимальной скоростью в 118 км/ч. Помнится, такая же цифра стояла в паспортной книжке дедушкиного “Запорожца” ЗАЗ-968М. BYD Flyer в минимальной комплектации стоит 5990 долл.
Первым в списке самых дешевых автомобилей, имеющих в своем арсенале хоть какие-то современные дополнительные опции, также идет BYD Flyer. За 6990 долл. у него появляются электростеклоподъемники, легкосплавные диски, центральный замок с пультом ДУ и магнитола с CD-проигрывателем. За доплату можно установить кондиционер.
В ситуации с Chery Sweet и Daewoo Matiz вышло так, что клон стал дороже оригинала. Chery Sweet сделан по образу и подобию корейского автомобиля, по такому случаю даже был громкий судебный процесс. Победа осталась за Chery. Однако покупатели в России предпочитают узбекский автомобиль Daewoo Matiz, годом рождения которого считается 1998-й. Дизайн для этого автомобиля был разработан в Италии. С 2001 года Matiz собирается в Узбекистане на заводе Uz-Daewoo. За сумму в 7400 долл. можно приобрести самую бедную комплектацию этого автомобиля. За блага цивилизации нужно существенно доплачивать. Относительно новенький Chery Sweet за 7640 долл. обладает более экономичным мотором и уже установленным в базе кондиционером.
Дизайн для этого автомобиля был разработан в Италии. С 2001 года Matiz собирается в Узбекистане на заводе Uz-Daewoo. За сумму в 7400 долл. можно приобрести самую бедную комплектацию этого автомобиля. За блага цивилизации нужно существенно доплачивать. Относительно новенький Chery Sweet за 7640 долл. обладает более экономичным мотором и уже установленным в базе кондиционером.
Hafei Brio – относительно новая модель, ее производят всего 2,5 года. Автомобиль начал свою жизнь вполне цивилизованно – в 2002 году по заказу китайцев один из грандов итальянского автомобильного дизайна, студия Pininfarina, разработала внешний вид небольшого и недорогого автомобильчика. Никакого клонирования, копирования и пародирования известных моделей. У Brio за 7690 долл. литровый, но маломощный (46 л.с.) моторчик. Разгоняется он так же вяло, как и другие конкуренты по цене, а в поворотах сильно кренится. Светлый салон из китайского пластика не понравился российским покупателям, и специально для России сделали салон из черных материалов.
Чуть дороже, чем самые дешевые китайские автомобили, сегодня стоит ВАЗ-2113. Рестайлинговая “восьмерка” посвежела, но потеряла целостность форм, присущих родоначальнику переднеприводных ВАЗов модели 2108 1984 года. За долгие годы эксплуатации этот автомобиль снискал славу машины с острым управлением и неплохим комфортом. За два десятка лет, что выпускается ВАЗ-2108, его конструкцию изучили все автосервисы страны, и дефицит на запчасти к нему давно канул в Лету. Несмотря на возраст, эта модель наиболее приспособлена к эксплуатации в суровых российских условиях. Пятидверная машина ВАЗ-2114 уже выходит за рамки 8 тысяч долларов.
| Модель: | ВАЗ-11116 (“Ока”) | ВАЗ-21053 | ВАЗ-21053 | BYD Flyer | |||||
| Год: | 1989 | 1980 | 1982 | 2001 | |||||
| Возраст: | 17 лет | 26 лет | 24 года | 5 лет | |||||
| Длина (мм): | 3200 | 4130 | 4145 | 3605 | |||||
| База (мм): | – | 2424 | 2424 | 2300 | |||||
Снар. масса (кг): масса (кг): | 665 | 1060 | 1060 | 720 | |||||
| Скорость (км/ч): | 150 | 150 | 150 | 118 | |||||
| Разгон до 100 км/ч (сек.): | 18 | 17 | 17 | 25 | |||||
| Расход (город, л/100 км): | 5,5 | 9,7 | 9,7 | – | |||||
| Расход (трасса, при 90 км/ч): | 4 | 8,5 | 8,5 | 4,4 | |||||
Объем двиг. (см³): (см³): | 993 | 1451 | 1451 | 796 | |||||
| Мощность двиг. (л.с.): | 53 | 76 | 76 | 40 | |||||
| Крут. момент (Нм): | 77 | 112 | 112 | 62 | |||||
| Цена ($): | 5170 | 5200 | 5650 | 5990 | |||||
| Производство: | Россия | Россия | Россия | Китай | |||||
| Модель: | Daewoo Matiz | Chery Sweet | Hafei Brio | ВАЗ-2113 | |||||
| Год: | 1998 | 2003 | 2004 | 1984 | |||||
| Возраст: | 8 лет | 3 года | 2 года | 22 года | |||||
| Длина (мм): | 3495 | 3550 | 3588 | 4122 | |||||
| База (мм): | 2340 | 2340 | 2335 | 2460 | |||||
Снар. масса (кг): масса (кг): | 778 | 880 | 920 | 975 | |||||
| Скорость (км/ч): | 144 | 148 | 120 | 165 | |||||
| Разгон до 100 км/ч (сек.): | 15,2 | 18,5 | – | – | |||||
| Расход (город, л/100 км): | 7,3 | – | 5,5 | 10,7 | |||||
| Расход (трасса, при 90 км/ч): | 6,3 | 4,5 | 4,5 | 6,5 | |||||
Объем двиг. (см³): (см³): | 796 | 812 | 1000 | 1499 | |||||
| Мощность двиг. (л.с.): | 54 | 51 | 46 | 56 | |||||
| Крут. момент (Нм): | 69 | 70 | 72 | 116 | |||||
| Цена ($): | 7400 | 7640 | 7690 | 7850 | |||||
| Производство: | Узбекистан | Китай | Китай | Россия | |||||
Кирилл Орлов
Сколько весит ока машина – АвтоТоп
Технические характеристики ВАЗ ОКА. Узнайте габариты, расход топлива ВАЗ ОКА, особенности двигателей, подвесок, кузовов и прочих технических характеристик автомобилей ВАЗ ОКА.
Узнайте габариты, расход топлива ВАЗ ОКА, особенности двигателей, подвесок, кузовов и прочих технических характеристик автомобилей ВАЗ ОКА.
Выберите модель ВАЗ ОКА:
Ответы
История ВАЗ ОКА
Микролитражная Ока остается самым дешевым отечественным легковым автомобилем. В свое время ее даже прочили на роль народного автомобиля и определяли местом ее производства гигантский промышленный комплекс в Елабуге, намереваясь покончить с многолетним автомобильный дефицитом. Но мечты и проекты так и ocтались неисполнимыми, а столь досаждавшая АвтоBA3y сборка «Оки» была в середине 1990-х окончательно передана на заводы СеАЗ (который вошел в состав АвтоВАЗа) и КамАЗ.
Идея сделать из «Оки» народный автомобиль с самого начала была утопией, ведь основное предназначение личного легкового автомобиля у нас в стране – это перевозка семьи по выходным на дачу и вывоз оттуда сельхозпродукции, обеспечить которой горожан власть была так же не в состоянии, как и легковыми автомобилями.
Но все меняется, и не всегда в худшую сторону. Да, народного автомобиля из Оки не получилось, но свое место под солнцем она заняла. Ведь более удобного для города, а тем паче дешевого отечественного автомобиля вы не найдете. Карбюраторный 649-кубовый 30-сильный двигатель – это половинка «вазовской» восьмерки, что видно по унификации важнейших узлов поршневой группы с 1,3-литровым двигателем ВАЗ-2108. Многие другие узлы взяты от модели 2105, например радиатор, кран и арматура отопления, что избавляет от многих проблем с запчастями, но не решает их совсем, так как некоторые детали (типа коленвала и т. п). оригинальны, а масштаб производства мал и цены соответственно могут быть и больше, чем у «жигулёвских» или «самарских». Вообще это правило, к сожалению, применимо к значительной доле запчастей в общем-то дешевой «Оки». Поэтому тщательно взвешивайте при покупке баланс цены и последующих затрат на ремонт микролитражки (возможно, капитальный).
В салоне легко размещаются четверо человек средней комплекции с поклажей, что довольно удивительно при таких малых габаритных размерах (3200х1430х1400 мм). Вместимость багажника при сложенных задних сиденьях (и не сильно отодвинутых передних) позволяет грузить не только мелкий скарб, но и достаточно крупные вещи типа 100-литрового холодильника или 25-дюймового телевизора. А вот прогулками на природу, да еще с полной загрузкой ие стоит злоупотреблять, так как проходимость довольно ограниченна (можно и глушитель оторвать) и пассивная безопасность далека от совершенства, зато управляемость на дороге довольно приличная и по энергоемкости подвески нет даже сравнения с импортными «одноклассниками», тем более подержанными.
Вместимость багажника при сложенных задних сиденьях (и не сильно отодвинутых передних) позволяет грузить не только мелкий скарб, но и достаточно крупные вещи типа 100-литрового холодильника или 25-дюймового телевизора. А вот прогулками на природу, да еще с полной загрузкой ие стоит злоупотреблять, так как проходимость довольно ограниченна (можно и глушитель оторвать) и пассивная безопасность далека от совершенства, зато управляемость на дороге довольно приличная и по энергоемкости подвески нет даже сравнения с импортными «одноклассниками», тем более подержанными.
В городе же «Ока» действительно очень юркий (использующий любую щель между стоящими машинами) и экономичный автомобильчик, и если его не перегружать и заезживать «до дыр», то вполне еще достойный. Однако возраст берет свое: шум и вибрация в салоне – вещи, кажется, неизбежные. И хотя посадка на передних сиденьях довольно свободная даже для людей высокого роста (почему тучные «новые русские» иногда покупают ее для поездок в гараж за «мерседесом»), три педали где-то сбоку (сцепление аккурат под правую ногу приходится!), торчащий, как палка, рычаг переключения передач и недостаток тепла для задних пассажиров от печки, комфорта ей в повседневной эксплуатации не добавляют.
Что, впрочем, для роли второго автомобиля в семье или первого в жизни (для бедных студентов) не большая помеха. Тем более что по ходу производства часть недостатков (бедной комплектации) была в начале 1990-х устранена: так, появился отопитель заднего стекла, в сиденьях – тканевые вставки, а багажник закрыли полочкой.
В 1996 году в производство запущена модель 11113 с 750-кубовым двигателем мощностью 36 л.с., которая в сравнении с предшественником более динамична и экономична (6 л бензина АИ-93 на 100 км в городском цикле) благодаря меньшей рабочей частоте вращения.
В 1999 г. предприятию удалось изготовить 28 тыс. микролитражек, а в 2000 г. рассчитывают довести выпуск до 32 тыс. единиц. Тем не менее количественный рост сдерживают ограниченные поставки с АвтоВАЗа силовых агрегатов, приводов и других комплектующих. Положение должно измениться после того, как производство двигателей в Тольятти достигнет 60 тыс. единиц (2000 г.). Также на ЗМА началась эксплуатация нового сварочного комплекса с годовой производительностью 44 тыс. кузовов.
кузовов.
В настоящее время подавляющая часть собираемых на конвейере микролитражек приходится на долю трехдверного хэтчбека ВАЗ-11113 «Ока». Находящийся в его моторном отсеке двухцилиндровый двигатель рабочим объемом 0,75 л мощностью 33 л.с. обеспечивает максимальную скорость 130 км/ч. Динамические качества модели улучшат, так как в 2001 г. на Авто-ВАЗе освоят выпуск 40-сильных двигателей, оснащенных системой впрыска топлива. Коробка передач – 4-ступенчатая, привод осуществляется на передние колеса. По заказу устанавливают измененную переднюю панель, правое наружное зеркало заднего вида, колеса из легкого сплава, более комфортабельные передние сиденья с подголовниками, улучшают отделку интерьера. Кузов могут окрасить эмалями типа «металлик».
Попытки радикально осовременить великовозрастную микролитражную «Оку» пока отложены. Дальше постройки опытных образцов дела не идут. Тем не менее автомобиль поставляется на экспорт, решаются вопросы об организации сборочных производств за рубежом. А в России некоторые фирмы активно занимаются тюнингом «Оки».
А в России некоторые фирмы активно занимаются тюнингом «Оки».
Компания «Ока-Тюнинг» из Набережных Челнов в течение ряда лет является деловым партнером завода микролитражных автомобилей (ЗМА) и Серпуховского автозавода (СеАЗ), поставляя обоим предприятиям передние панели для заказных модификаций автомобиля «Ока». Вместе с тем фирма сама выпускает и реализует модернизированные ею микролитражки. Они отличаются накладными порогами, делающими боковины более динамичными, изящным спойлером над задней дверью, декоративными накладками на блоки наружных световых приборов, оригинальной формой отверстий для забора воздуха, дополнительными катафотами. Полностью обновлен салон автомобиля. Передняя панель гармонична и рациональна. Контрольно-измерительные приборы, заимствованные у ВАЗ-2106, удачно сгруппированы в едином корпусе. Иное расположение вентиляционных отверстий способствует лучшему распределению воздушных потоков. Появился вместительный перчаточный ящик с крышкой и запором. По-иному решено оформление внутренних панелей дверей. Теперь на них имеются удобные поручни. Передние и задние сиденья с более качественной двухцветной обивкой лучше отвечают требованиям эргономики.
Теперь на них имеются удобные поручни. Передние и задние сиденья с более качественной двухцветной обивкой лучше отвечают требованиям эргономики.
Специалисты дизайн-ателье «Карди» создали тюнинговый вариант популярной микролитражки «Ока», названный «Динара». Утонченная эстетика кузова достигнута благодаря переднему бамперу оригинальной формы, который выполнен заодно с облицовкой радиатора. Отверстия для забора воздуха хорошо сочетаются со стандартными указателями поворота и подвешенными снизу дополнительными фарами. Капот получил выштамповку, рельефно выделяющую среднюю часть. Задний бампер плавно сопряжен с поверхностью третьей двери. Снизу смонтированы трубчатые подножки, а задняя дверь увенчана спойлером. Для окраски кузова «Динары» используется широкая гамма цветов. Салон полностью обновлен, включая новые отделочные материалы. Передняя панель с контрольными приборами, клавишами и переключателями от ВАЗ-2106. Вместо стандартных использованы литые 13-дюймовые колеса. Серийный двигатель может быть заменен силовым агрегатом мощностью 40 или 63 л. с.
с.
ВАЗ-1111 Ока «ЭЛЕКТРО» – четырехместный электромобиль, созданный на базе серийно выпускаемого автомобиля Ваз 1111 «Ока». Применение злектрического привода делает его нетоксичным и бесшумным городским транмпортом.
Раздельная тормозна система, возможность рекупиративного торможения электродвигателем, травмобезопасное рулевое управление – соответствуют современным требованиям безопасности, предъявляемым к конструкции автомобиля. Быстродействующая эффективная защита при коротком замыкании полностью исключает поражение электрическим током, делая электромобиль совершенно безопасным в эксплуатации.
Ока «ЭЛЕКТРО» – трехдверный седан. Передние крылья выполнены съемными. Для посадки на заднее сидение спинки передних сидений наклоняются вперед. Заднее сиденье может складываться, освобождая дополнительно место для багажа.
Сейчас на АвтоВАЗе активно ведутся работы по созданию принципиально новой «Оки» – ВАЗ 1121 Ока-М, производство которой планируется развернуть в 2005-2006 годах. На сей раз это будет уже не глубокая модернизация модели, а полностью новый автомобиль, для которого разрабатывается не только новый кузов, но и шасси.
На сей раз это будет уже не глубокая модернизация модели, а полностью новый автомобиль, для которого разрабатывается не только новый кузов, но и шасси.
По предварительной информации уже к 2005 году будет закончен весь комплекс испытаний, и в том же году автомобиль по планам должен встать на конвейер. ВАЗ 1121 Ока-М как и нынешнюю модель, будут выпускать на двух заводах – СеАЗ и ЗМА. По оценкам специалистов, объем продаж автомобилей такого класса может составлять в России 150-200 тысяч штук.
Новая ВАЗ 1121 Ока-М станет по всем параметрам больше сегодняшней: на 100 мм длиннее, на 100 мм шире и на 100 мм выше. Вырастет мощность двигателя. Сейчас на «Оку» устанавливают мотор объемом 0,749 см/куб мощностью 33 л.с. Предварительно ВАЗ 1121 Ока-М будет оснащатся одним из двух двигателей: 33-сильным или более мощным украинским МеМЗ объемом 1,1 литра мощностью 49 л.с.
По предварительной информации ВАЗ 1121 Ока-М будет иметь все же 3-дверный кузов, хотя на макетах видно, что отрабавтывалась и 5-дверная версия. В настоящий момент доступна лишь фотография макета будущего автомобиля, однако увидеть концепцию новинки и очень предварительно оценить дизайн можно по представленному несколько лет назад концепту «Карат». Кстати самые первые концептуальные образцы «Оки-2» (первоначальное условное название) были выполнены еще в стиле модного тогда биодизайна и у ВАЗа не нашлось тогда денег на освоение такой модели, поэтому в дальнейшем концепция новой модели была полностью пересмотрена.
В настоящий момент доступна лишь фотография макета будущего автомобиля, однако увидеть концепцию новинки и очень предварительно оценить дизайн можно по представленному несколько лет назад концепту «Карат». Кстати самые первые концептуальные образцы «Оки-2» (первоначальное условное название) были выполнены еще в стиле модного тогда биодизайна и у ВАЗа не нашлось тогда денег на освоение такой модели, поэтому в дальнейшем концепция новой модели была полностью пересмотрена.
Двигатель ока: описание, характеристики и тюнинг
В настоящее время можно встретить огромное количество автомобилей и моторов. Все они отличаются не только стоимостью, но и техническими характеристиками. В данной статье мы поговорим про довольно необычный двигатель Ока, распространенный в советское и настоящее время.
Технические характеристики
Отправить на email
| Модель | ВАЗ – 1111 |
| Тип | Четырехтактный, бензиновый, карбюраторный |
| Число и расположение цилиндров | Два цилиндра расположены в ряд |
| Диаметр цилиндра и ход поршня | Диаметр равен 76 мм, ход поршня равен 71 мм |
| Объем двигателей, л | 0. 649 649 |
| Степень сжатия | 9.9 |
| Номинальная мощность двигателей при 5600 оборотах | 21,5 кВт, или 29,3 лошадиные силы |
| Максимальная мощность при 3200 оборотах | 44 кВт, или 4,5 лошадиные силы |
| Октановое число | От 92 до 95 |
| Система зажигания | Бесконтактная |
| Система подачи топлива | Карбюратор |
| Свечи зажигания | А17ДВРМ, FE65CPR |
| Вес, кг | 63.5 |
| Расход в городском цикле, л | 6 |
| Расход в смешанном цикле, л | 3.9 |
Двигатель устанавливается на ВАЗ – 1111, ВАЗ — 1113
Описание
Представленная модель относится к типу четырехтактных моторов на карбюраторной основе.
Его отличительная черта состоит в том, что распределительный вал находится в верхней части двигателя от Оки. Не все современные и прошедшие свою жизнь силовые агрегаты оснащены такой изюминкой.
Стоит отметить, что все цилиндры имеют рядное расположение, что позволяет получать очень хороший технический потенциал. Конструкторы автомобиля Ока взяли одну половину от всеми известного ВАЗ 21083 и сделали ее основой. Из этого следует, что некоторые детали и механизмы не отличаются от своего старшего собрата.
Что касается системы охлаждения, то тут стоит жидкостная циркуляция закрытого типа. Поэтому двигатель от Оки сможет работать в любых погодных условиях, что очень хорошо. Он не боится лютых морозов и жары. Благодаря комбинированной системе смазки все детали и механизмы будут работать практически без сбоев.
Конструкция
По сути, это стандартный мотор от ВАЗ 21083, который пользуется большой популярностью. Двигатель Ока немного видоизменен и имеет вес в два раза меньший. За счет этого удалось получить небольшой расход топлива и высокий КПД.
В основе лежит простой четырехтактный силовой агрегат, работающий на бензиновом топливе. К главной особенности относится верхнее расположение распределительного вала и рядное расположение цилиндров. Благодаря этому удалось получить высокую мощность.
Благодаря этому удалось получить высокую мощность.
В стандартной комплектации водитель получает комбинированную систему смазки:
За счет этого все детали и механизмы получат свою порцию масла и не будут выходить из строя. Что касается системы охлаждения, то тут все стандартно. Конструкторы установили жидкостную систему, которая циркулирует жидкость по всему контуру. Благодаря этому двигатель Ока не будет перегреваться под солнцем.
Сейчас можно встретить всего две модификации двигателя:
Обслуживание
В данный раздел входит не только замена масла, но и регулировка зазоров в клапанном механизме.
Первым делом стоит рассказать про самое простое, замена моторного масла.Для этого вам потребуются следующие инструменты:
- ключ на 17;
- небольшая емкость под отработавшую жидкость;
- новое масло;
- отвертка.
Перед началом работ нужно заглушить мотор автомобиля и установить опоры под колеса. Это нужно для безопасности, чтобы во время работ не приключилось неожиданностей.
Замена масла
Если вы хотите полностью заменить тип масла (если тип масла остается прежним, то ниже указанные действия проводить не нужно):
- сначала промойте всю систему промывочным маслом;
- Для этого нужно немного влить жидкость до нижней отметки и запустить двигатель от Оки. Специалисты рекомендуют дать ему поработать в течение 10 минут. За это время специальная жидкость пройдет несколько раз по всей системе и соберет весь шлак.
- После этого можно слить ее и заменить масляный фильтр. Это нужно для того, чтобы новое масло не получило неизвестных веществ во время прохождения фильтра.
- Заливать моторное масло следует до верхней отметки, до максимума. Далее ждем около 10 минут и при необходимости доливаем моторную жидкость.
- Повернуть крышку горловины и снять ее;
- Убрать пробку сливного отверстия под двигателем автомобиля. Это нужно для того, чтобы старое масло слилось в специальную емкость;
- Завернуть пробку обратно;
- Стоит помнить, что во время сливания масло очень горячее.
 Далее можно отвернуть масляный фильтр и убрать его в сторону. Больше он не понадобится;
Далее можно отвернуть масляный фильтр и убрать его в сторону. Больше он не понадобится; - Для откручивания фильтра лучше использовать специальный ключ;
- Заполняем новое масло в полость фильтра до середины;
- Не нужно забывать про смазывание уплотнительного кольца, которое располагается на фильтре;
- Заливаем новое моторное масло в горловину до максимума и заворачиваем крышку;
- Запускаем мотор и даем ему поработать в течение нескольких секунд. За это время можно проверить наличие подтеканий и уровень масла. При необходимости его можно увеличить.
Регулировка зазора
Для компенсации расширения деталей во время работы конструкторы предусмотрели специальный зазор между стержнем клапана и самим кулачком. При увеличении зазора клапана могут не открываться, а при уменьшении не закрываться. За этим нужно следить практически каждый день.
Проверять зазор нужно только на холодном двигателе, когда все детали находятся в стандартном состоянии. Минимальный зазор впускного клапана должен быть 0,2 миллиметра, а выпускного 0,35 миллиметра. Если этого не соблюдать, то двигатель Ока будет работать нестабильно, с некоторым шумом и свистом.
Минимальный зазор впускного клапана должен быть 0,2 миллиметра, а выпускного 0,35 миллиметра. Если этого не соблюдать, то двигатель Ока будет работать нестабильно, с некоторым шумом и свистом.
Клапана нужно считать от ремня распределительного вала: 1-й и 4-й клапаны выпускные, а 2-й и 3-й впускные. Стоит отметить, что порядок регулировки не важен. То есть проводить регулировку можно в любой последовательности.
Для данной работы вам потребуется ключ на 10, набор отверток, щупов и приспособления для регулировки зазоров. Если прокрутить коленчатый вал не получается, то можно использовать следующий совет. Включите четвертую передачу и очень медленно прокатите транспортное средство до того момента, пока кулачок не займет свое положение.
- Снять воздушный фильтр;
- Прикрыть карбюратор специальной тряпкой, чтобы ничего туда не попадало;
- Открутить крышку головки блока цилиндров;
- Несколько раз провернуть коленчатый вал специальным ключом. Для упрощения данного действия можно включить передачу и вывернуть свечи зажигания;
- Метки шкива генератора и передней крышки должны совпасть.
 Если этого не случилось, значит, вы не до конца провернули колесо. Во время данного действия можно приступать к регулировке клапанов автомобиля. Стоит обратить свое внимание на то, что на крышке находятся две метки. Ориентироваться надо только на длинную метку;
Если этого не случилось, значит, вы не до конца провернули колесо. Во время данного действия можно приступать к регулировке клапанов автомобиля. Стоит обратить свое внимание на то, что на крышке находятся две метки. Ориентироваться надо только на длинную метку; - Далее следует измерить тепловой зазор при помощи плоского щупа. Эти значения можно записать в тетрадку и сравнить с номинальными в специальном руководстве;
- Прикрепить на крышку специальное устройство;
- Надеть шайбы и провернуть их;
- Нажать на рычаг и утопить толкатель. Вставить фиксатор под распределительный вал так, чтобы выступ зафиксировал толкатель;
- Далее можно поддеть регулировочную шайбу и вынуть ее. Записать толщину шайбы;
- Рассчитываем толщину новой шайбы по формуле: H = B + A — C,где H — толщина новой шайбы;B — толщина старой шайбы;A — значение измеренного зазора;C — номинальный зазор;
- Устанавливаем ее в толкатель и производим действия в обратном порядке;
- Проворачиваем коленчатый вал на 360 градусов и регулируем тепловой зазор;
- Устанавливаем детали в обратном порядке, чтобы двигатель Оки был в прежнем состоянии.

Неисправности
| Перегрев силового агрегата | Плохая система охлаждения |
| Дрожит рычаг коробки | Износ подушек крепления мотора |
| Стук авто Ока с китайским двигателем | Неотрегулированные клапана |
Тюнинг
Многие водители хотят сделать из своего железного коня настоящего монстра. Это касается и небольшого автомобиля Ока.
Если вы хотите увеличить мощность, то можно сделать тюнинг двигателя Оки.
Для этого нужен специальный инструмент, мастерская и опыт:
- Первым делом можно заменить головку блока цилиндров, воздушный фильтр, купить новый распределительный вал и карбюратор. По сути, это самый минимум, что можно сделать на первом этапе.
- Вы знаете, что у автомобиля Ока объем двигателя не очень большой, и это никак не исправить. Конечно, можно попробовать установить другой двигатель, но это очень сложно и трудоемко.
 К менее затратному относится чип тюнинг, который позволяет немного увеличить крутящий момент и компрессию. К тому же уменьшается расход топлива, что очень хорошо.
К менее затратному относится чип тюнинг, который позволяет немного увеличить крутящий момент и компрессию. К тому же уменьшается расход топлива, что очень хорошо. - Многие специалисты рекомендуют отшлифовать коллектор, толкатель и распределительный вал. Данные действия относятся к основному тюнингу двигателя Ока. При помощи этого можно не только увеличить мощность, но и сделать работу двигателя бесперебойной.
- Многие водители задают вопрос: какие двигатели можно поставить на автомобиль Ока? И ответ очень простой. Только стандартный или немного измененный от ВАЗ 21083.
Технические характеристики СеАЗ (Ока) 11116, 1111, 11113 — Remont-Avtovaz.ru
- Короткая предистория
- Идентификация автомобиля
- Габаритные параметры
- Двигатели и трансмиссии
- Подвеска, рулевое управление, тормозная система
- Видео — Тест-драйв Ока «грузовая» СеАЗ
Предистория
В 1988 году с конвейера «АвтоВАЗа» сошел автомобиль особо малого класса ВАЗ «Ока». В дальнейшем производство было передано на другие заводы, основным же стал Серпуховский завод, с соответствующим обозначением модели – СеАЗ. Но при этом название модели – «Ока», оставалось неизменным.
В дальнейшем производство было передано на другие заводы, основным же стал Серпуховский завод, с соответствующим обозначением модели – СеАЗ. Но при этом название модели – «Ока», оставалось неизменным.
Первая модель являлась базовой и имела она индекс Ока 1111. В дальнейшем появилось еще несколько моделей, получивших улучшенные технические характеристики и другие индексы. Самыми массовыми из них стали модели с индексами 1113 и 11116.
Производство данного автомобиля остановлено в 2008 году. Являясь практически самым малым автомобилем отечественного производства, ВАЗ 1111 технические характеристики имел вполне неплохие для своего класса.
Идентификация автомобиля
Как и все авто, «Ока» имела идентификационные номера, наносившиеся на кузов и двигатель. Номер кузова дополнительно являлся идентификационным обозначением самого автомобиля.
На кузов автомобиля идентификационный номер наносился в трех местах, что исключало возможность его подделки. Первый номер был нанесен на специальную табличку, закрепленную спереди на кузове, в подкапотном пространстве. Эта табличка также несла информацию в виде кода о заводе-изготовителе, на нее наносился индекс модели и модельный год выпуска.
Эта табличка также несла информацию в виде кода о заводе-изготовителе, на нее наносился индекс модели и модельный год выпуска.
Идентификационный номер дополнительно наносился на кузов под решетку воздухозаборника, расположенного возле лобового стекла.
Третий номер был нанесен внутри салона, на поперечине пола багажника.
У двигателя же идентификационный номер был только в одном месте – на блоке цилиндров передней части, рядом со вторым цилиндром.
Габаритные параметры
Все версии авто имели несущий трехдверный кузов, и рассчитан был автомобиль на 4 пассажиров. Несмотря на то, что выпускались три разные модели, габаритные показатели у них были идентичны. Длина авто составляла всего 3200 мм, при ширине – 1420 мм, и почти с такой же высотой – 1400 мм. При этом колесная база у «Оки» составляла 2180 мм. Клиренс у всех моделей был равен 150 мм. А вот по снаряженной массе они отличались, у 1111 масса составляла 640 кг, у 1113 – 645 кг, а у 11116 – 665 кг. Сказывалось использование двигателей с разными конструктивными особенностями.
Сказывалось использование двигателей с разными конструктивными особенностями.
Двигатели и трансмиссии
Двигатель «Ока» характеристики имел разные, в зависимости от модели. Ока 1111 и 1113 оснащались двухцилиндровыми силовыми агрегатами с синхронным ходом поршней и уравновешивающим механизмом. Система питания – карбюраторная, охлаждение – жидкостное, а система зажигания – электронная бесконтактная, с использованием датчика Холла.
У Ока 1111 технические характеристики мотора сводились к общему объему камер сгорания в 0,649 литра и мощности 29,3 л.с. По сути, это была половина 1,3-литрового мотора модели ВАЗ-2108.
У СеАЗ-1113 объем был больше – 0,75 литра, мощность тоже была выше – 33,0 л.с.
СеАЗ 11116 технические характеристики имел несколько иные. Эта модель, появившаяся позже всех, комплектовалась китайской силовой установкой с 3 цилиндрами. Рабочий объем этого агрегата составлял уже 1,0 литр, а мощность, которую он выдавал, составляла 53 л.с.
Все автомобили были переднеприводными. На версиях 1111 и 1113 использовалась 4-ступенчатая коробка передач, оснащенная синхронизаторами каждой передачи. Скоростной максимум Ока 1111 составлял 115 км/ч, а 1113 – 130 км/ч.
На версиях 1111 и 1113 использовалась 4-ступенчатая коробка передач, оснащенная синхронизаторами каждой передачи. Скоростной максимум Ока 1111 составлял 115 км/ч, а 1113 – 130 км/ч.
Китайский мотор версии 11116 работал в паре уже с 5-ступенчатой коробкой. Это позволяло разогнать авто до 150 км/ч.
Подвеска, рулевое управление, тормозная система
Передок авто оснащался подвеской типа МакФерсон, с телескопическими амортизаторами, поперечными рычагами и поперечным стабилизатором устойчивости. Сзади же применялась подвеска, состоящая из амортизаторов, винтовых пружин, продольных рычагов и поперечной балки.
Рулевое управление «Ока»…
травмобезопасное, сделанное по типу «шестерня-рейка». Передача усилия от него на колеса производилось двумя рулевыми тягами.
Передние колеса оснащались дисковыми тормозами…
суппорт был подвижным. Регулировка зазоров между суппортом и колодками осуществлялось автоматически.
На задних колесах механизмы были барабанные, между колодками и барабаном регулировка зазора выполнялась автоматически. Привод рабочих тормозов – гидравлический, стояночный же имел тросовый привод.
Привод рабочих тормозов – гидравлический, стояночный же имел тросовый привод.
Статья в тему — Устройство двигателя ОКИ ( СеАЗ)
Бортовая электрическая сеть…
авто была однопроводной, отрицательным полюсом был сам кузов. Номинальное напряжение в сети составляло 12 В. Напряжение в сети поддерживалось двумя источниками – АКБ и генератором.
В целом, автомобиль «Ока» являлся очень интересным, однако нерентабельность производства и неконкурентоспособность с зарубежными авто такого класса привела к сворачиванию его производства.
Видео — Тест-драйв Ока «грузовая» СеАЗ
Технические характеристики Oka | Vaz
Трёхдверный хэтчбэк ВАЗ 1111 Ока за двадцать лет своей истории – с 1988 до 2008 года – стал одним из самых популярных моделей «народного автомобиля». И в самом деле, сравнительно небольшая мощность и исключительно маленькие размеры (особо малый класс) позволили снизить стоимость автомобиля Ока до пределов, вполне доступных для самой широкой аудитории.
Дизайн кузова Оки сильно напоминал японскую модель Daihatsu Mira, вышедшую на рынок в 1985 году, однако отечественные инженеры уверяли, что эта похожесть – только видимость. Технически же модель ВАЗ 1111 Ока была вполне самостоятельной новинкой советского автопрома.
А дешевизна обслуживания ВАЗ 1111 Ока позволила сделать автомобиль основой для различных модификаций – в частности, пикапов и снегоходов.
Первоначально автомобили ВАЗ 1111 Ока оснащались двухцилиндровым мотором, объёмом в 0,65 литра и мощностью всего 29,7 лошадиных сил («половинка» двигателя от «восьмёрки», объём которого составлял 1,3 литра). Однако вскоре после выхода автомобиля в массовое производство у Оки появились более мощные двигатели – на 33 и даже 53 лошадиные силы. Первый из них также представлял собой «половинку» полуторалитрового двигателя модификации ВАЗ 2108 (3), двухцилиндровый, объёмом в 0,75 литра.
Уже в 2005 году, несмотря на уникальность Оки в российском сегменте А, морально устаревшая модель стала проигрывать на рынках, в особенности – зарубежных. Адаптацию же двигателя Оки к экологическим нормам Евро-2 на ВАЗе сочли нецелесообразной. Именно поэтому Ока так и не получила инжекторный двигатель взамен своих карбюраторных моделей.
Адаптацию же двигателя Оки к экологическим нормам Евро-2 на ВАЗе сочли нецелесообразной. Именно поэтому Ока так и не получила инжекторный двигатель взамен своих карбюраторных моделей.
Лишь в короткий период с 2006 по 2008 производимые на СеАЗе модификации автомобиля (СеАЗ-11116) получили литровые 3-цилиндровые инжекторные двигатели китайского производства, создаваемые для всё тех же автомобилей Daihatsu. Там же производились и пикап-версии Оки – СеАЗ-11116-50 и СеАЗ-11116-60. Эти автомобили были адаптированы и к нормам Евро-2, и даже к Евро-3, однако также скоро уступили рынок за нерентабельностью.
Технические характеристики автомобиля Ока
Наименование Автомобиль
ВАЗ-1111 ВАЗ-1113
Количество мест 4
Количество мест при сложенном заднем сиденьи 2
Полезная масса, кг: 340
Масса перевозимого груза, кг
при водителе и трех пассажирах
при водителе и пассажире
40
190
Масса снаряженного автомобиля (полностью заправленного
и снаряженного, но без полезной нагрузки), кг 635 645
Просвет автомобиля с полной массой при статическом
радиусе шин 237 мм, не менее, мм
до подрамник
до поддона картера двигателя
150
170
Внешний наименьший радиус поворота по оси следа переднего колеса, не более, м. 4,6
4,6
Максимальная скорость, км/ч
с полной массой
с водителем и одним пассажиром
115
120 130
135
Время разгона с места с переключением передач
до скорости 100 км/ч, с
с полной массой
с водителем и одним пассажиром
36
30 24
20
Максимальный подъем, преодолеваемый автомобилем с полной массой, на участке сухого ровного и твердого грунта без разгона на первой передаче (для .обкатанного автомобиля с приработанным двигателем, протяженность подъема не менее двойной длины автомобиля), % 30
Тормозной путь автомобиля с наибольшей нагрузкой со скорости 80 км/ч на горизонтальном участке сухого ровного асфальтированного шоссе, не более, м
при использовании рабочей тормозной системы
при использовании одного из контуров рабочей системы
43,2
93,3
Двигатель
Модель ВАЗ-1111 ВАЗ-1113
Тип четырехтактный, бензиновый, карбюраторный
Число и расположение цилиндров 2, в ряд
Диаметр цилиндра и ход поршня, мм 76х71 82×71
Рабочий объем, л 0,649 0,749
Степень сжатия 9,9
Номинальная мощность при частоте вращения коленчатого вала 5600 об/мин по ГОСТ 14846Ч81 (нетто), кВт (л. с.) 21,5(29,3) 24,3 (33)
с.) 21,5(29,3) 24,3 (33)
Максимальный крутящий момент при частоте вращения коленчатого вала 3200 об/мин по ГОСТ 14846Ч81 (нетто), Н.м (кгс.м) 44(4,5) 50 (5,1)
Трансмиссия
Сцепление однодисковое, сухое, с центральной нажимной пружиной
Привод выключения сцепления — тросовый, беззазорный
Коробка передач четырехступенчатая, с синхронизаторами на всех передачах
переднего хода. Главная передача цилиндрическая, косозубая
Дифференциал конический, двухсателлитный
Привод передних колес валы с шарнирами равных угловых скоростей
Ходовая часть
Передняя подвеска независимая, с телескопическими гидравлическими
амортизаторными стойками, с винтовыми цилиндрическими
пружинами, нижними поперечными рычагами
с растяжками и стабилизатором поперечной устойчивости
Задняя подвеска на продольных, взаимосвязанных рычагах, с винтовыми
цилиндрическими пружинами и телескопическими,
гидравлическими амортизаторами двухстороннего действия
Колеса дисковые, штампованные размер обода 4B-12h3
для камерных и бескамерных шин
Шины радиальные, низкопрофильные,
камерные или бескамерные.
Размер — 135/80 R12
Рулевое управление
Тип травмобезопасное
Рулевой механизм шестерня-рейка
Рулевой привод две тяги с шарнирным креплением к торцам рейки
и шарнирным соединением с поворотными рычагами
Тормоза
Рабочая тормозная система: передний тормозной механизм дисковый, с подвижным суппортом и автоматической
регулировкой зазора между диском и колодками
Задний тормозной механизм барабанный, с самоустанавливающимися колодками,
с автоматической или ручной регулировкой зазора между
колодками и барабаном
Тормозной привод гидравлический, двухконтурный с диагональным разделением
контуров, с вакуумным усилителем и регулятором давления
Стояночный тормоз ручной, с тросовым приводом на колодки тормозных
механизмов задних колес
Схема электрооборудования однопроводная, отрицательный полюс источников питания
соединен с массой. Номинальное напряжение 12В.
Аккумуляторная батарея 6СТ-36А, зарядом 130000 Кл (36 А.ч)
Генератор 37. 3701, переменного тока, со встроенным выпрямителем
3701, переменного тока, со встроенным выпрямителем
на кремниевых диодах и электронным регулятором
напряжения. Ток отдачи 55 А при 5000 об/мин’
Стартер 39.3708, дистанционного управления,
с электромагнитным включением
и муфтой свободного хода
Кузов
Модель ВАЗ-1111а
Тип цельнометаллический, несущий, трехдверный
Передаточные числа 5-и ступенчатой коробки передач:
первая передача … 3,7
вторая передача … 2,06
третья передача … 1,27
четвертая передача … 0,90
задний ход … 3,67
главная передача … 4,54 (4,1)
Основные данные для регулировок и контроля:
Параметр Значение Допуск
Зазоры в механизме привода клапанов на холодном двигателе, мм:
для впускных клапанов
для выпускных клапанов
+0,05. -0,05
+ 0,05. -0.05
Минимальная частота вращения коленчатого вала, мин 820.900
Давление масла в системе смазки двигателя при температуре масла 85С
и частоте вращения коленчатого вала 5600 об/мин, не менее, МПа (кгс/см*2) 0,45 (4,5)
Минимальное давление масла в системе смазки двигателя при температуре
масла 85С и частоте вращения коленчатого вала 820.3 1,078 — 1,085
Зазор между электродами свечи зажигания, мм 0,7 — 0,8
Начальный угол опережения зажигания до ВМТ, град
ВАЗ 1111
ВАЗ 11113
1
4
Полный ход педали сцепления, мм 110 5
Свободный ход педали тормоза при неработающем двигателе, мм 3.5
Минимальная допустимая толщина накладок для колодока
передних и задних тормозов, мм 1,5
Уровень жидкости в бачке гидропривода тормозов приа
установленной крышке до нижней кромки
Максимальный уклон на сухом твердом грунте, на котором
автомобиль с полной нагрузкой удерживается неограниченное
время стояночным тормозом при перемещении рычага
на 4.5 зубцов сектора, % 30
Свободный ход рулевого колеса а положении, соответствующем движению по прямой,
не более, град 5
Схождение передних колес для обкатанного автомобиляа
под нагрузкой 2200 Н (225 кгс), мм 0 +1
-1
Развал передних колес для обкатанного автомобиля под
нагрузкой 2200 Н (225 кгс), град 0 +30
-30
То же при замере между ободом и вертикалью, мм -3.2) 0.18(1,8)
| 1. Снять отрицательный провод
с аккумулятора.
Предупреждение На автомобилях
с кондиционером, элементы системы кондиционирования воздуха могут
затруднять выполнение работ на двигателе, и не всегда возможно отвинтить
болты и переместить элементы системы в сторону из-за недостаточной
длины их шлангов. В таком случае, система кондиционирования должна
быть разряжена дилером Форда или специалистом по кондиционерам. |
|
| 2. Снять защиту картера и брызговик. | |
| 3. Снять решетку радиатора. | |
| 4. Отсоединить трос открытия капота от механизма закрывания капота и извлечь из фиксаторов. | |
| 5. Снять капот, предварительно отметив положение петель, для облегчения повторной установки и отсоединив трубку омывателя ветрового стекла. | |
| 6. Слить охлаждающую жидкость из системы охлаждения, для чего под сливную пробку радиатора подставить соответствующий контейнер и открыть сливную пробку, расположенную в нижней части радиатора. Для ускорения слива охлаждающей жидкости открыть пробку расширительного бачка. Для слива охлаждающей жидкости из блока цилиндров отвинтить дренажный болт, расположенный в блоке цилиндров. | |
| 7. Для обеспечения дополнительного рабочего места, снять радиатор. | |
| 8. Отсоединить шланги охлаждающей жидкости от корпуса водяного насоса с левой стороны двигателя и головки блока цилиндра. | |
| 9. Ослабить хомуты и снять патрубки
и шланги системы охлаждения: – верхний и нижний патрубки радиатора; – шланги расширительного бачка; – шланги системы отопления салона от головки блока цилиндров и насоса системы охлаждения; – корпуса автоматического пускового устройства карбюратора. |
|
| 10. Слить моторное масло и снять фильтр. | |
| 11. Отвинтить четыре болта крепления верхней передней поперечины, два болта с двух сторон вверху и два нижних болта крепления опоры поперечины и снять ее вместе с радиатором. | |
| 12. Снять воздушный фильтр. | |
| 13. Снять шланг сапуна c крышки головки блока цилиндров и отвинтить болт, крепящий подвеску поддержки шланга к левой передней стороне головки блока цилиндра. | |
| 14. Снять с топливного насоса подводящий топливный шланг и заглушить его пробкой. | |
| 15. Отсоединить вакуумную трубку усилителя тормозов от впускного коллектора. | |
| 16. Отключить электрические провода
и снять разъемы со следующих агрегатов и датчиков: – генератора; – стартера; – вентилятора радиатора; – датчика оборотов и положения коленчатого вала; – датчика температуры; – датчика температуры системы управления двигателем; – датчика давления масла; – распределителя зажигания; – катушки зажигания; – узлов системы впрыска топлива и топливных форсунок; – датчика положения дроссельной заслонки; – датчика температуры топлива; – подогревателя автоматической воздушной заслонки; – термодатчика радиатора. |
|
| 17. Снять с распределителя зажигания распределитель высокого напряжения вместе с проводами. | |
| 18. Отсоединить трос акселератора. | |
| 19. Отсоединить от карбюратора топливные и вакуумные шланги, а также электрические провода. | |
| 20. Снять клиновой ремень и вспомогательный насос системы рулевого управления (если присутствует в данной версии). Подвесить вспомогательный насос к кузову автомобиля при помощи мягкой проволоки, при этом не отсоединять шланги от насоса. | |
| 21. В салоне автомобиля установить брус под педалью сцепления так, чтобы зафиксировать ее в верхнем положении, для исключения срабатывания автоматического регулятора натяжения троса сцепления. | |
| 22. Снять трос сцепления. | |
| 23. На моделях с кондиционированием воздуха, отвинтить 4 болта крепления компрессора кондиционера (указаны стрелками на рисунке (вид снизу)) от подвески установки и переместить компрессор от двигателя. | |
| 24. Проверить, что рулевое колесо установлено в положении движения прямо вперед и краской или маркером отметить положение нижнего хомута промежуточного вала и шестерни рулевой передачи, затем отсоединить промежуточный вал от рулевой передачи (болт крепления указан стрелкой). | |
| 25. Отвинтить переднюю приемную трубу от выпускного коллектора. | |
| 26. Отвинтить хомуты крепления стабилизатора и снять стабилизатор. | |
| 27. На автомобилях с автоматической коробкой передач отвинтить преобразователь крутящего момента от приводного диска. | |
| 28. Установить таль для подъема двигателя и приподнять двигатель настолько, чтобы вес двигателя воспринимался талью, а затем отвинтить держатели двигателя. | |
| 29. Отвинтить болты крепления картера сцепления. | |
| 30. Отсоединить электрические провода и снять стартер. | |
| 31. Отсоединить от двигателя провод массы. | |
| 32. Снять держатель, расположенный между двигателем и коробкой передач. | |
| 33. Отсоединить трубки тормозной системы от передней подвески. | |
| 34. Подпереть коробку передач при помощи соответствующего подъемника и широкой доски и немного приподнять коробку передач. | |
| 35. Соблюдая осторожность, грузоподъемным механизмом приподнять двигатель и отделить от него коробку передач. | |
| 36. Приподнимая двигатель, одновременно сместить его вперед и, поворачивая его в горизонтальной плоскости, извлечь вверх. | |
Ока 1111 и 11113 расход топлива на 100 км
Ока – автомобиль, выпускаемый на российских заводах ВАЗ, СеАЗ и КамАЗ. История его разработки начинается еще в 1970 годы на Серпуховском автомобильном заводе, а сам выпуск Оки стартовал только в 1988 году. За двадцатилетний период производства в общей сложности было выпущено около 700 тысяч автомобилей.
Содержание страницы
Ока 0.7 МТ
Норма расхода топлива на 100 км
С самого начала автомобиль выпускался с двигателем объемом 0.7 литра, мощность которого составляет 30 лошадиных сил. Максимальная скорость, которую мог развить 30-сильный, равна 120 км/ч, а его расход топлива – 6.5 л в городском цикле и 5.5 л на трассе.
Отзывы владельцев о расходе бензина
- Сергей, Ростов. Lada (ВАЗ) 1111 (Oka) 0.7 МТ 2005 г. Это была моя первая машина, сейчас езжу на тринадцатой модели. Машина неплохая тем, что неприхотливая в ремонте и расход экономичный. На 100 км в среднем около 6 литров уходит. За все время чистил только карбюратор и все. Автомобиль очень хорошо подойдет для новичков.
- Максим, Москва. Машина так себе, так как Ока предназначена для перевоза только одного человека, то есть просто себя покатать. Качество запчастей на низшем уровне. Приходилось в своем ВАЗе 1993 года полностью все перебирать. Единственное что хорошо это расход — в городе не больше 7 литров, на трассе 5.5-6 литров.
- Тарас, Уфа. Машину брал с рук. Сразу же после покупки Оки 2000 года выпуска поменял тормозные колодки и топливный фильтр. В целом неплохая тачка, особенно хорошо подходит для езды по городу, с парковкой проблем не возникает. Из-за малого веса на трассе машину швыряет во все стороны. Расход топлива 6-6.5 литра.
- Георгий, Рязань. Езжу на Ладе Ока 1994 года давно, и меня до сих пор все в ней устраивает. Отрабатывает на все 100 процентов. Самое главное — это следить за автомобилем и менять расходники. Двигатель 0.7 МТ резвенький, особенно при старте. Расход тоже отличный – всего-навсего 6.5 литра в городе и 5.5 литра на трассе.
- Ирина, Тамбов. Лада 1111 0.7 МТ 2007 г. Машинка неплохая и маневренная. Для обучения езды и что где находится под капотом в самый раз. Расход в среднем 6 литров. Минус в том, что года через полтора машина начинает сыпаться как не следи за ней. Очень часто рвутся патрубки от печки, плюс каждые 6 месяцев приходится подваривать пороги.
Оka 0.8 МТ
Официальная информация
В 1995 году на смену 0.7-литрового пришел новый 2-цилиндровый двигатель с объемом 0.8 литра. Мощность такого агрегата составляет 33 лошадиные силы, а максимальная скорость — 130 км/ч. При этом расход топлива в городском цикле 7.4 л, на трассе — 5 л.
Реальный расход топлива
- Владимир, Питер. Lada (ВАЗ) 11113 (Oka) 0.8 МТ 2000 г. В машине, конечно, комфорт отсутствует, но по своим тяговым качествам очень даже нормальная. Двигатель работает неплохо. Масла много не расходует, топлива тоже. На 100 км выходит в районе 6-6.5 литра. Большим минусом Лады является слабенький кузов, который плохо противостоит коррозии.
- Андрей, Орел. В своей Оке 2004 года вижу только плюсы. Очень компактный автомобиль – можно проехать где угодно. Мотор 0.8 МТ расходует топлива мало: в городе около 7 литров, на трассе при 70 км/ч 5 литров. Несмотря на такой маленький расход, двигатель сохраняет неплохую динамику, двух человек тянет без проблем.
- Евгений, Владимир. Покупал Оку в 2007 году с незначительным пробегом. Машина неплохая, но сейчас уже пересел на другую. После доработки подвески управление было отличное. Проездил на ней 4 года, после чего кузов стал сыпаться, слабоватый. Большое достоинство данной машины это двигатель 0.8 МТ, разгоняется неплохо и на трассе держит скорость в 120 км/ч. Расход 7-7.5 литра.
- Кирилл, Курск. Lada (ВАЗ) 11113 (Oka) 0.8 МТ 2002 г. Несмотря на многие недостатки машины, она все-таки неплохая. Салон, конечно, без каких-либо удобств. Ломается постоянно, но ремонт стоит сущие копейки. Очень часто лопались бампера. Расход топлива небольшой — где-то 7.5 литра в городе, 5 литров на трассе.
- Дмитрий, Ульяновск. Машину брал с рук (модель 1997 года выпуска). Очень долго простояла в гараже. Поменял все резиновые детали, так как поусыхались. Еще пришлось сменить проводку – сильно плавилась. Сейчас езжу, в принципе все пока что устраивает. Расход бензина в городе 7.3-7.5 литра, на трассе при 100 км/ч 5.5 литра.
Ока 1.0 МТ
Информация от производителя
Наряду с двигателями 0.7 и 0.8 литра автомобиль выпускался с мотором объемом 1.0 литра. При этом мощность силового агрегата значительно больше двух предыдущих – 53 л.с. Максимальная скорость — 150 км/ч, а расход топлива в городе 6.2 л, на трассе — 4.5 л.
Владельцы о расходе
- Алексей, Симферополь. Несмотря на ежедневную эксплуатацию автомобиля, а это поездки не меньше 500 км в день, автомобиль Лада (Ока) 1999 года выпуска 1.0 МТ и сегодня работает как часы. Если и возникали какие-то проблемы, то все очень быстро решалось и исправлялось. В общем, автомобиль классный, особенно расход — в городе около 6.5 литра и на трассе до 5 литров.
- Юрий, Москва. Oka 1.0 МТ 2000 года. До недавних пор являлся владельцем данного «чуда». Машина, одним словом, ведро на колесах. Я удивляюсь, что многие еще находят плюсы в этой «коляске» и пишут хорошие отзывы. Положительных сторон в ней нет. Да и расход для 1.0-литрового движка почти в 7 литров это слишком много.
- Александр, Калуга. Средства при покупке авто у меня были ограничены. А так как покупал свой первый автомобиль, знакомые посоветовали Оку 1992 года с мотором 1.0 МТ. В целом она, наверное, для этого и предназначена, чтобы неопытные в вождении тренировались, так как на ней можно только ездить по городу и все. К тому же расход в пределах 6 литров на 100 км.
- Василий, Киров. Предпочитаю машины отечественного производства. Вот решил взять себе Оку 99-года с движком 1.0. Кроме нее есть еще ГАЗ-3110. Машина хороша тем, что можно ремонтировать без особо каких проблем, но поиск запчастей составляет некоторую трудность. Расход в городе 6.5 литра, на трассе 4.5-5 литров.
- Дмитрий, Самара. Lada (ВАЗ) 1111 (Oka) 1.1 МТ 1998 г. На отметке пробега в 140 тысяч км заклинил двигатель. После ремонта ездит как новая, даже немного добавилось мощи. Минус в том, что особо не погоняешь на ней, слишком малый вес и из-за этого плохая управляемость. Зимой плохо заводилась, поменял свечи все теперь нормально. Расход бензина в среднем 5 литров, что очень неплохо.
Идентификация нового антагониста тромбоцитов, который связывается с CLEC-2 и подавляет вызванную подопланином агрегацию тромбоцитов и метастазирование рака.
ВВЕДЕНИЕ
Метастазирование — очень сложный процесс и основная причина смерти, связанной с раком. Большинство раковых клеток, попадающих в кровоток, быстро устраняются за счет высокого сдвигового кровотока и иммунной системы хозяина. Менее 0,1% раковых клеток, вытесненных из первичного очага опухоли, выживают в кровотоке и вызывают метастазы [1].Взаимодействие между циркулирующими опухолевыми клетками и тромбоцитами часто приводит к индуцированной опухолевыми клетками агрегации тромбоцитов (TCIPA), которая способствует гематогенному метастазированию опухоли [1, 2]. Был разработан ряд средств против TCIPA для блокирования метастазов рака [3–6]. Аспирин, апираза, тканевой ингибитор металлопротеиназы-4, BM-567, XV454 и абциксимаб входят в число агентов, которые ингибируют взаимодействия опухолевых клеток с тромбоцитами и метастазы рака [7–11]. Лечение онкологических больных этими агентами обычно приводит к увеличению риска кровотечений, поскольку большинство анти-TCIPA агентов действуют на гемостатические белки тромбоцитов [4, 5, 12, 13].Разработка агентов против TCIPA, которые не мешают физиологическому гемостазу, имеет решающее значение для их использования в качестве части противоракового терапевтического режима.
Подопланин (PDPN) является одним из наиболее часто активируемых генов при плоскоклеточной карциноме, опухолях центральной нервной системы и зародышевой неоплазии [14, 15]. PDPN индуцирует агрегацию тромбоцитов путем связывания и активации лектин-подобного рецептора 2 C-типа (CLEC-2) [16, 17], что приводит к фосфорилированию тирозина киназ семейства Src, Syk и фосфолипазы C гамма 2 (PLCγ2) [18– 22].PDPN усиливает образование метастатических очагов и прогрессирование опухоли, не влияя на рост опухоли в исследованиях на животных [16]. Метастатическая активность рака и частота эмболии опухолевых клеток в микроциркуляторном русле легких коррелируют с активностью PDPN по агрегации тромбоцитов [16, 23-25]. Специфическое ингибирование передачи сигналов CLEC-2 не должно влиять на физиологический гемостаз, поскольку тромбоциты с дефицитом CLEC-2 нормально реагируют на тромбоцитарные агонисты коллагена, АДФ, U46619 и пептид рецептора 4, активируемого протеазой [26].Это указывает на то, что PDPN-индуцированное взаимодействие тромбоцитов с опухолевыми клетками является потенциальной мишенью для разработки режима антиметастазирования.
Это исследование включает исследование нового небольшого синтезированного соединения 2CP, производного 4- O -бензоил-3-метокси-бета-нитростирола (BMNS), которое специфически связывается с CLEC-2 и ингибирует PDPN-индуцированный TCIPA. . 2CP вызывает и увеличивает терапевтическую эффективность противораковых препаратов, не влияя на нормальный гемостаз в модели ксенотрансплантата мыши.Также определены новые сигнальные белки, передающие PDPN-активируемую сигнализацию CLEC-2. Обсуждается значение этих результатов для разработки режима лечения рака.
РЕЗУЛЬТАТЫ
2CP селективно ингибирует PDPN-индуцированную агрегацию тромбоцитов
BMNS, неселективный ингибитор агонист-индуцированной агрегации тромбоцитов [27–31], был использован в качестве ведущего соединения для химического синтеза BMNS-родственных производных RX1, RX41 и 2CP (рисунок 1A, дополнительный метод и дополнительный рисунок S1).Было проанализировано влияние этих соединений на агонистическую агрегацию тромбоцитов. BMNS и RX41 неизбирательно ингибировали агрегацию тромбоцитов, индуцированную всеми агонистами, исследованными в этом исследовании, включая PDPN, тромбин, коллаген, ADP и U46619 (рис. 1B). RX1 обладал умеренным ингибированием агрегации тромбоцитов, стимулированной PDPN или коллагеном, и сильным ингибированием агрегации тромбоцитов, стимулированной тромбином, ADP или U46619. 2CP ингибировал PDPN-индуцированную агрегацию тромбоцитов с IC50, эквивалентным 12.1 ± 4,8 мкМ (рис. 1В и таблица 1), но мало влиял на агрегацию тромбоцитов, индуцированную родоцитином, агонистом CLEC-2, или тромбином, коллагеном, АДФ или U46619 (рис. 1В и 1С). Все значения IC50 2CP для этих агонистов были> 100 мкМ (таблица 1). Эти результаты показывают, что 2CP избирательно ингибирует PDPN-индуцированную агрегацию тромбоцитов.
Фигура 1: Селективное ингибирование PDPN-индуцированной агрегации тромбоцитов с помощью 2CP. A. Химическая структура производных BMNS. B. Промытые тромбоциты предварительно инкубировали с контрольным носителем ДМСО или указанными соединениями (20 мкМ) при 37 ° C в течение 3 минут и затем стимулировали PDPN (8 мкг / мл), тромбином (0,1 Ед / мл). , коллаген (2 мкг / мл), ADP (20 мкМ) и U46619 (2 мкМ) соответственно. Показаны характерные следы агрегации тромбоцитов. C. Промытые тромбоциты (1 × 10 9 / мл) предварительно инкубировали с контрольным носителем ДМСО или 2CP (20 мкМ) при 37 ° C в течение 3 минут и затем стимулировали указанными агонистами CLEC-2 ( родоцитин: 2 мкг / мл; PDPN: 8 мкг / мл).Агрегацию тромбоцитов регистрировали агрегометром тромбоцитов, и показаны репрезентативные следы агрегации тромбоцитов для всего не менее четырех независимых экспериментов. Стрелки указывают точку добавленного агониста.
Таблица 1: Влияние 2CP на агрегацию тромбоцитов, индуцированную различными агонистами
Соединение | Конц. (мкМ) | Светопропускание (% ингибирования контроля) a | ||||
|---|---|---|---|---|---|---|
PDPN | Тромбин | Коллаген | ADP
| U46619 | ||
Контроль | 0 | 79.2 ± 1,9 | 78,3 ± 0,9 | 74,0 ± 1,2 | 34,6 ± 2,9 | 76,1 ± 1,7 |
2CP | 100 900 ± | ** (89) | 61,0 ± 4,3 ** (22) | 60,0 ± 5,1 * (18) | 26,5 ± 2,9 (23) | 65,6 ± 2,6 ** (13 ) |
20 | 19.8 ± 5,4 ** (75) | 75,7 ± 0,9 (3,3) | 69,3 ± 2,4 (6,3) | 31,6 ± 2,2 (8,6) | 72,6 ± 2,3 (4,5) | |
5 | 59,8 ± 8,9 (24) | 76,3 ± 1,0 (2,5) | 70,7 ± 1,4 (4,4) | 31,5 ± 3,2 (8,9) | 74,3 ± 1,4 (2,3) | |
BMNS | 20 | 5.8 ± 1,2 ** (92) | 2,6 ± 2,6 ** (96) | 0,1 ± 0,1 ** (99) | 1,1 ± 2,4 ** (96) | 0,5 ± 0,3 ** (99) |
a Показан процент пропускания света в конце анализа агрегации. Данные представляют собой среднее значение ± стандартное отклонение ( n ± 4).
* p <0,05 ** p <0,01 по сравнению с соответствующим контролем агониста.В скобках указан процент ингибирования светопропускания по сравнению с контролем.
2CP ингибирует PDPN-индуцированный TCIPA
C6 / клетки легких использовали в качестве модели раковых клеток для выяснения того, способен ли 2CP ингибировать PDPN-индуцированный TCIPA. Это было основано на выводах о том, что клетки C6 / легких экспрессируют высокие уровни PDPN и индуцируют агрегацию тромбоцитов (рис. 2A) и образование агрегатов опухолевые клетки-тромбоциты (рис. 2B). Напротив, родительские клетки C6 / LG не экспрессировали PDPN и не были способны индуцировать агрегацию тромбоцитов и образовывать агрегаты опухолевые клетки-тромбоциты (рис. 2A и 2B).Экспрессия короткой шпилечной РНК PDPN в клетках C6 / легких устраняет агрегацию тромбоцитов, вызванную C6 / клетками легких (рис. 2C), что означает, что PDPN является ключевой молекулой, опосредующей TCIPA.
Рисунок 2: 2CP ингибирует PDPN-опосредованный TCIPA. A. Экспрессию белков PDPN в указанных клеточных линиях определяли вестерн-блоттингом с использованием антитела против PDPN (вставленная панель). Экспрессию β-актина использовали для контроля равной нагрузки белка. Клетки (1,5 × 10 6 ) из указанных клеточных линий добавляли к суспензии промытых тромбоцитов человека (1 × 10 9 / мл) для стимуляции агрегации тромбоцитов.Агрегацию тромбоцитов, индуцированную опухолевыми клетками, измеряли и регистрировали с помощью агрегометра. Показаны характерные следы агрегации тромбоцитов. B. Тромбоциты, меченные зеленым кальцеином-AM (1 × 10 9 / мл, зеленые), инкубировали с помеченными оранжевым / красным клетками кальцеина-AM (1,5 × 10 6 , красный) в агрегометре. Затем реакционные смеси помещали на предметное стекло для анализа с помощью флуоресцентной микроскопии. Представлены типичные флуоресцентные изображения, демонстрирующие взаимодействие между опухолевыми клетками и тромбоцитами.Масштабная линейка = 20 мкм. C. Экспрессию белка PDPN в указанных клеточных линиях определяли вестерн-блоттингом с использованием антитела против PDPN (левая панель). Экспрессию β-актина использовали для контроля равной нагрузки белка. Активность этих сублиний по индукции агрегации тромбоцитов оценивали с помощью анализов TCIPA. Показаны характерные следы агрегации тромбоцитов (правая панель). Стрелки указывают точку добавления ячеек. D. Клетки из указанных клеточных линий добавляли к промытым тромбоцитам с предварительной инкубацией с 2CP (20 мкМ) или без нее.Показаны характерные следы агрегации тромбоцитов (левая панель) и мутность реакций (центральная панель). Время достижения 50% максимальной агрегации было определено как время агрегации, которое показано в виде прямоугольника с усами (от минимального до максимального) (правая панель). Значение устанавливается на 1000 секунд, если агрегация тромбоцитов не наблюдалась. ** P <0,01 по сравнению с обработкой носителем. E – F. Тромбоциты и клетки C6 / легких обрабатывали указанными концентрациями 2CP и измеряли активности ЛДГ и каспазы 3/7.Данные представляют собой среднее значение ± стандартное отклонение от трех до шести независимых экспериментов.
АнализыTCIPA проводили путем инкубации клеток C6 / легких с тромбоцитами человека в присутствии или в отсутствие 2CP. 2CP ингибировал агрегацию тромбоцитов, вызванную C6 / клетками легких (рис. 2D, левая панель), и вызывал помутнение реакционной смеси (рис. 2D, центральная панель). Время достижения 50% агрегации для контрольной группы и группы, получавшей 2CP, составляло 496,5 ± 65,4 с ( n = 41) и 910,6 ± 123,1 с ( n = 39), соответственно (Рисунок 2D, правая панель, p <0.01). Анализы высвобождения лактозодегидрогеназы (ЛДГ) и активности каспазы 3/7 показали, что 2CP не вызывает цитотоксических или апоптотических эффектов в тромбоцитах и опухолевых клетках. Это означает, что клеточный стресс или гибель клеток не учитывают ингибирующую активность 2CP в отношении TCIPA, индуцированную клетками C6 / легких (рис. 2E и рис. 2F). 2CP также ингибировал TCIPA, индуцированный клеточными линиями остеосаркомы человека HOS и MG63, которые экспрессируют высокие уровни PDPN (фиг. 3). Эти результаты показывают, что 2CP ингибирует PDPN-опосредованный TCIPA.
Рисунок 3: 2CP ингибирует агрегацию тромбоцитов, индуцированную клетками остеосаркомы MG-63 и HOS. Экспрессию PDPN в клетках остеосаркомы MG-63 и HOS определяли с помощью вестерн-блоттинга (верхняя панель) и проточной цитометрии (средняя панель). Для анализов TCIPA клетки MG-63 (2 × 10 6 ) или HOS (1,5 × 10 6 ) добавляли в промытые тромбоциты (1 × 10 9 / мл) с предварительной обработкой 2CP ( 20 мкМ). Агрегацию тромбоцитов регистрировали агрегометром в течение 20 мин (нижняя панель).
Влияние 2CP на время кровотечения из хвоста мыши и легочные метастазы в модели ксенотрансплантата мыши
Влияние 2CP на функцию тромбоцитов in vivo оценивали путем внутривенной доставки 2CP мышам B6 с последующим анализом времени кровотечения из хвоста ( Рисунок 4A). В этом анализе использовалась доза 3,5 мг / кг, что эквивалентно 5-кратному Kd (24,5 ± 3,7 мкМ) для связывания PDPN и CLEC-2 [18]. Не было разницы во времени кровотечения между контролем (87.4 ± 8,1 с, n = 23) и обработанных 2CP (85,6 ± 7,1 с, n = 21) мышей ( p = 0,87). Время кровотечения при внутривенной доставке гепарина и низкомолекулярного гепарина (НМГ) также сравнивали с 2CP. При дозировке 2 мг / кг (300 МЕ / кг для гепарина и 200 МЕ / кг для НМГ), которая обычно использовалась для ингибирования метастазов рака в модели ксенотрансплантата мыши [32–35], время кровотечения из хвоста составляло 333,5 ± 11,7 с ( n = 11) и 169,9 ± 30,8 с ( n = 11) для гепарина и НМГ соответственно.Время кровотечения у животных было значительно продлено гепарином и НМГ по сравнению с контролем и 2CP ( p <0,01). Таким образом, 2CP не оказывает вредного воздействия на нормальный гемостаз.
Рисунок 4: Влияние 2CP на время кровотечения из хвоста и легочные метастазы у мышей. A. Мышам внутривенно вводили контрольный носитель ДМСО, 2CP (3,5 мг / кг), гепарин (2 мг / кг) или LMWH (2 мг / кг) с последующим измерением времени кровотечения из хвоста мыши.Время кровотечения наносили на график и выражали как среднее значение ± стандартное отклонение. Время кровотечения более 360 секунд было установлено как 360 секунд. B. График экспериментальных протоколов модели легочных метастазов у мышей. График введения и терапевтическая эффективность для 2CP и CDDP (2,5 мг / кг) анализировали в указанные моменты времени. C – E. Были показаны репрезентативные изображения биолюминесценции для роста опухоли (панель C, слева) и количественно определены (панель C, справа). Репрезентативные изображения срезов легких с метастатическими очагами (окраска гематоксилином и эозином) анализировали и количественно оценивали с помощью программного обеспечения Zeiss Axiovision.Стрелками указаны очаги метастазирования. (100-кратное увеличение, масштабная линейка = 100 мкм). Соответствующая метастатическая область выражалась в процентах от всей области легкого (панель D). Регистрировали массу тела и рассчитывали процент потери массы тела на 21 день после инокуляции опухоли. Данные представляют собой среднее значение ± стандартное отклонение. от трех до пяти независимых экспериментов (панель E). * P <0,05 и ** P <0,01 по сравнению с контрольной обработкой. n.s., не имеет значения.
Модель ксенотрансплантата мыши была затем использована для исследования того, оказывает ли 2CP какое-либо влияние на легочные метастазы клеток C6 / легких. 2CP вводили голым мышам в присутствии или в отсутствие противоракового агента цисплатина (CDDP) с использованием протокола, показанного на рисунке 4B. Образование опухоли контролировали с помощью анализа биолюминесцентной визуализации и гистопатологического исследования легочной ткани на 15 день после внутривенной доставки мышам клеток С6 / легких. Оба 2CP ( p <0.05) и CDDP ( p <0,01) снижали сигнал биолюминесценции и количество метастатических очагов (рис. 4C и 4D). Комбинированное лечение мышей 2CP и CDDP дополнительно уменьшало сигнал биолюминесценции и количество очагов метастазов опухоли по сравнению с мышами, получавшими только CDDP ( p <0,01). Площадь метастазов для контроля, 2CP, CDDP и комбинированного лечения 2CP и CDDP составляла 54,4% ± 1,8% ( n = 17), 46,7% ± 2,1% ( n = 17), 35.6% ± 3,3% ( n = 16) и 19,1% ± 1,8% ( n = 15) исследуемой площади поверхности соответственно (рис. 4D).
Потеря массы тела для каждой группы лечения, зарегистрированная на 21 день после инокуляции раковых клеток, показала, что 2CP сам по себе не влиял на животных (фиг. 4E). В сочетании с CDDP 2CP снижал степень потери массы тела одновременно с уменьшением опухолевой нагрузки (рис. 4E, p <0,01). В соответствии с этими результатами, только 2CP не увеличивал продолжительность жизни мышей, в то время как CDDP увеличивал выживаемость животных ( p = 0.015) по сравнению с контрольной группой (Рисунок 5). Комбинированное лечение 2CP и CDDP вызывало дальнейшее увеличение продолжительности жизни мышей по сравнению с контролем ( p <0,001) или лечением только CDDP ( p = 0,008). В отличие от эффектов in vivo 2CP на метастазы клеток C6 / легких и выживаемость животных, в клетках C6 / LG, которые не экспрессировали PDPN, 2CP не продлевал выживаемость животных и не уменьшал образование метастатических очагов в легких ( Дополнительный рисунок S2).Эти данные предполагают, что 2CP специфически подавляет рост и метастазы раковых клеток, экспрессирующих PDPN.
Фигура 5: Комбинация 2CP с CDDP увеличивает время выживания экспериментальных животных. Мышам внутривенно вводили клетки C6 / Lung (1,75 × 10 5 ) с последующей обработкой 2CP ( n = 21), CDDP (2,5 мг / кг, n = 22), 2CP + CDDP. ( n = 23) или управление транспортным средством ( n = 21), как описано в материалах и методах.Выживаемость животных регистрировалась, и данные анализировались с использованием кривой выживаемости Каплана-Мейера и критерия логарифмического ранга.
Мы также выясняем, подавляет ли 2CP метастазирование в легких и увеличивает ли выживаемость животных, когда инъекция опухоли предшествовала доставке 2CP мышам (дополнительная фигура S3A). Наши данные показали, что комбинированное лечение мышей 2CP и CDDP немного уменьшило опухолевую нагрузку ( p = 0,07), но не повлияло на процент потери веса тела по сравнению с одним CDDP (дополнительные рисунки S3B и S3C).2CP был эффективным в продлении выживаемости животных. Фракция выживания мышей составляла 0%, 14,2%, 28,4% и 56,8% для лечения контрольным носителем, 2CP, CDDP и комбинированного лечения 2CP и CDDP, соответственно (дополнительный рисунок S3D). Эти данные подтверждают мнение о том, что 2CP эффективен в продлении выживаемости животных с опухолями, экспрессирующими PDPN.
2CP выявляет новую передачу сигналов CLEC-2 тромбоцитов
Эффект 2CP на PDPN- и родоцитин-индуцированное фосфорилирование тирозина тромбоцитов было проанализировано и сравнено для определения молекулярной основы 2CP на ингибировании PDPN-индуцированной агрегации тромбоцитов и TCIPA ( Рисунок 6А).И PDPN, и родоцитин индуцировали фосфорилирование тирозина тромбоцитарных белков. В соответствии с дифференциальными эффектами 2CP на PDPN- и родоцитин-индуцированную агрегацию тромбоцитов (рис. 1C), только фосфорилирование, индуцированное PDPN, подавлялось 2CP (рис. 6A). Когда были проанализированы отдельные сигнальные белки ниже CLEC-2 (фиг. 6B), как PDPN, так и родоцитин индуцировали фосфорилирование PLCγ2 (Y1217), SLP76 (Y145) и Syk (Y525 / 526). Другие Syk-активированные сигнальные белки, включая Akt1 (S473), PKCμ (S748), p38 (Y180 / Y182) и цитозольную фосфолипазу A2 (cPLA2-S505), также фосфорилировались после стимуляции тромбоцитов PDPN и родоцитином.2CP ингибировал PDPN-индуцированное, но не родоцитин-индуцированное фосфорилирование этих белков. С другой стороны, фосфорилирование киназы 1 пируватдегидрогеназы (PDK1-S241) снижалось под действием PDPN и родоцитина с 2CP, обращая вспять эффекты PDPN, но не родоцитина. Эти находки показывают, что 2CP избирательно подавляет PDPN-индуцированную передачу сигналов CLEC-2, несмотря на тот факт, что PDPN и родоцитин разделяют одни и те же сигнальные белки ниже CLEC-2.
Фигура 6: 2CP селективно ингибирует PDPN-, но не родоцитин-индуцированную передачу сигналов тромбоцитов. A – B. Промытые тромбоциты (1 × 10 9 / мл) предварительно инкубировали с 2CP (20 мкМ) или без него при 37 ° C в течение 3 мин, а затем стимулировали PDPN (8 мкг / мл) или родоцитином (2 мкг / мл). мл). Лизаты цельных клеток получали из тромбоцитов, стимулированных агонистами, и фракционировали с помощью SDS-PAGE. Фосфорилирование белков детектировали с помощью антител против фосфотирозина (4G10) или фосфоспецифических антител к указанным белкам. Сигналы и уровни относительного фосфорилирования указанных белков измеряли и количественно оценивали с помощью программного обеспечения Image J (NIH).Данные представляют собой среднее значение ± стандартное отклонение. от четырех до восьми независимых экспериментов. ** P <0,01 по сравнению с контрольной обработкой.
Нацелен ли 2CP непосредственно на протеинкиназы, которые находятся ниже по ходу активации CLEC-2 или связаны с активацией тромбоцитов, было дополнительно проанализировано с помощью анализов активности протеинкиназы in vitro . 2CP не влиял на активность всех 25 проанализированных протеинкиназ (дополнительная таблица S1). Эти данные исключают основные протеинкиназы передачи сигналов тромбоцитов как прямые мишени для 2CP.
Прямое взаимодействие 2CP с CLEC-2
Было выполнено компьютерное молекулярное моделирование, чтобы выяснить, связывает ли 2CP напрямую CLEC-2. Докинг-анализ показал, что 2CP связывается с аминокислотными остатками Asn105, Arg107, Phe 116, Arg118 и Arg157 CLEC-2, образуя водородные связи у атомов кислорода боковой цепи (рис. 7A). Анализ поверхностного плазмонного резонанса (SPR) проводили, чтобы выяснить, связывает ли 2CP напрямую CLEC-2. Чип биосенсора был покрыт рекомбинантным CLEC-2, и через чип пропускали различные концентрации 2CP.Повышение концентрации 2CP вызывает усиление связывания с CLEC-2. Рассчитанная аффинность связывания была эквивалентна 33,2 ± 1,9 мкМ (фиг. 7B). Эти результаты проиллюстрировали прямое взаимодействие между 2CP и CLEC-2. Кроме того, мы отметили, что сайты связывания 2CP и PDPN на CLEC-2 перекрываются при наложении кристаллических структур PDPN и CLEC-2 с док-моделью 2CP и CLEC-2 (рис. 7C). Данные также предполагают, что 2CP ингибирует передачу сигналов CLEC-2, препятствуя связыванию PDPN и CLEC-2.
Рис. 7: Молекулярный докинг и анализ SPR показывают взаимодействие 2CP с CLEC-2. A. Моделирование молекулярного стыковки 2CP с мономерным CLEC-2 было выполнено с использованием программы Discovery Studio (Accelrys, San Diego, CA). 2CP показан в виде шара и клюшки с серыми углами. Ключевые остатки CLEC-2 показаны в виде палочек с серыми атомами углерода (слева) или розовыми кружками (справа). Взаимодействия водородных связей показаны пунктирными красными линиями (слева) или синими / розовыми линиями. B. Различные концентрации 2CP вводили на поверхности, связанные с CLEC-2, создавая зависимый от концентрации сигнал.Сенсограммы типичных экспериментов по связыванию на основе равновесия после вычитания фонового отклика от контрольной поверхности показаны на левой панели. График реакции равновесного связывания из сенсограмм в зависимости от концентрации 2CP показан на правой панели. Кривая лучше всего соответствует экспериментальным данным с расчетным сродством связывания 33,2 ± 1,9 мкМ. Результаты представляют четыре независимых эксперимента. C. Подопланин и 2CP совместно используют перекрывающийся сайт связывания, присутствующий в CLEC-2.Иллюстрация была создана путем наложения изображений кристаллической структуры связывания CLEC-2 (синий) -PDPN (желтый), полученных из базы данных PDB (ID = 3WSR), и моделирования молекулярного стыковки 2CP (модель шара и палки с серыми атомами углерода. ) с мономерным CLEC-2. Показаны ключевые аминокислоты (Asn 105, Arg107, Phe116, Arg118 и Arg157), участвующие в связывании 2CP (серые пунктирные линии представляют водородные связи). Модель показывает вышележащий PDPN и 2CP-связывающий карман в CLEC-2.
ОБСУЖДЕНИЕ
TCIPA играет ключевую роль в метастазах рака [1, 4, 5, 16, 36–38].Это исследование показало, что 2CP является первым синтетическим соединением, которое связывается с CLEC-2 и ингибирует PDPN-индуцированный TCIPA без вредного воздействия на физиологический гемостаз. 2CP в сочетании с противоопухолевым средством облегчает метастазы в легких у экспериментальных животных и является многообещающим средством для лечения метастазов рака.
Помимо участия в TCIPA, PDPN активируется на переднем конце инвазивного края опухолевой массы и участвует в коллективном процессе клеточной инвазии независимо от неэпителиально-мезенхимального перехода раковых клеток [23, 39].Экспрессия подопланина в клетках MDCK и клетках плоскоклеточного эпителия полости рта увеличивает миграцию отдельных клеток после потери экспрессии E-кадгерина [40]. Исследования in vitro и in vivo с использованием клеточных линий различного происхождения рака подтверждают мнение о том, что PDPN имеет решающее значение для роста рака и метастазирования, несмотря на то, что неизвестны молекулярные игроки и механизм, который определяет решение, какой из двух паттерны вторжения активируются PDPN [16, 24, 41].Клинические исследования раковых тканей человека показывают, что экспрессия PDPN связана с агрессивными фенотипами рака человека и является плохим прогностическим маркером при раке пищевода, полости рта, легких и шейки матки [15, 39, 42–46]. Таким образом, PDPN является важным кандидатом для разработки противоопухолевой терапии.
Для противораковой терапии были разработаны различные подходы к предотвращению взаимодействий PDPN / CLEC-2. Многообещающие результаты использования антител против PDPN были продемонстрированы в нескольких доклинических исследованиях.Антитела против PDPN NZ-1 и NZ-8 индуцировали зависимую от антител клеточную цитотоксичность и комплемент-зависимую цитотоксичность в устранении раковых клеток и подавлении TCIPA, роста опухоли и метастазов опухоли [47–49]. Химерные антитела против PDPN ChMS-1 и hP2–0 были разработаны для предотвращения роста рака и метастазов [25, 50]. Антитела против PDPN NZ-1, которые либо радиоактивно мечены N-сукцинимидил-4-гуанидинометил-3- [ 131 I] иодобензоатом, либо слитые с экзотоксином A Pseudomonas, несущим C-концевой пептид KDEL, продемонстрировали значительное подавление опухолевого роста злокачественных опухолей. глиома [51, 52].Стоимость производства антител и потенциальный иммунный ответ, связанный с доставкой антител, ограничивают прогресс в клиническом использовании этих агентов. 2CP имеет потенциал для преодоления затрат, связанных с использованием антител против PDPN, потому что его простой химический синтез и его низкая молекулярная масса позволяют его массовое производство. Основываясь на текущем исследовании и концепции рационального дизайна лекарств, можно синтезировать ряд производных 2CP для дальнейшего улучшения ингибирующей эффективности 2CP в отношении индуцированной PDPN агрегации тромбоцитов, связанной с ростом рака и метастазами.
2CP сам по себе не так эффективен, как совместное лечение 2CP с CDDP в экспериментальной модели метастазов. Это объясняется тем, что иммунитет хозяина или цитотоксические агенты необходимы для удаления раковых клеток из кровотока и эффективного подавления метастазов [5, 25, 52, 53]. 2CP не цитотоксичен и не убивает раковые клетки. Вместо этого 2CP подавляет TCIPA и снижает скорость и соотношение эмболизации опухоли к удаленным органам. Вмешательства в действие TCIPA недостаточно для подавления роста опухолевых клеток и предотвращения образования метастатических очагов.Это мнение согласуется с наблюдением, что большинство средств против TCIPA являются цитостатическими, но не цитотоксическими [4]. Таким образом, 2CP можно рассматривать в качестве адъювантного агента для лечения рака в присутствии противораковых цитотоксических режимов.
Молекулярная основа 2CP в ингибировании PDPN-индуцированной агрегации тромбоцитов и TCIPA также рассматривается в этом исследовании. Исходное соединение 2CP является ингибитором протеинкиназы [30, 31]. Хотя фосфорилирование / активация сигнальных белков CLEC-2, таких как Syk, PLCγ и SLP76, все подавляется в тромбоцитах, обработанных 2CP, 2CP, по-видимому, не играет прямой роли в качестве ингибитора протеинкиназы.Это подтверждается открытием, что 2CP не влияет на активность in vitro протеинкиназ, которые находятся ниже передачи сигналов CLEC-2 или связаны с активацией тромбоцитов. Дифференциальные эффекты 2CP на агрегацию тромбоцитов и фосфорилатон / активацию белков, индуцированные PDPN и родоцитином, которые разделяют большую часть сигнальных путей CLEC-2, также свидетельствуют против действия 2CP как протеинкиназы или ингибитора сигнального белка в тромбоцитах. Возникает вопрос: как 2CP специфически ингибирует PDPN-, но не родоцитин-индуцированную агрегацию тромбоцитов? Были предложены отчетливые связывающие свойства CLEC-2 с PDPN и родоцитином, несмотря на то, что одни и те же аминокислотные остатки Arg107, Arg118, Arg152 и Arg157 CLEC-2 вносят вклад во взаимодействие как с PDPN, так и с родоцитином [54].Различные сайты Arg118 в CLEC-2 участвуют в связывании O-гликана PDPN и C-концевого Tyr136 родоцитина. 2CP взаимодействует с аминокислотными остатками Asn105, Arg107, Phe116, Arg118 и Arg157 CLEC-2. Эти результаты предполагают, что 2CP взаимодействует с ключевыми аминокислотными остатками (Arg 107, Arg 118 и Arg 157), участвующими в связывании PDPN / CLEC-2 [54]. Следовательно, 2CP, вероятно, интегрируется в тот же карман связывания или пространственное расположение PDPN и препятствует связыванию PDPN, но не родоцитина, с CLEC-2.Эта модель связывания может объяснить причину специфического ингибирования PDPN-индуцированной агрегации тромбоцитов и TCIPA с помощью 2CP. Альтернативно, дифференциальный эффект 2CP на PDPN- и родоцитин-индуцированную активацию CLEC-2 можно объяснить разным сродством связывания CLEC-2 с PDPN и CLEC-2 с родоцитином, что эквивалентно 24,5 мкМ и 1 мкМ соответственно [18, 55]. 2CP, вероятно, более эффективно конкурирует во взаимодействии CLEC-2 с PDPN, чем во взаимодействии с родоцитином.Обе модели объясняют специфическое ингибирование PDPN-индуцированной агрегации тромбоцитов и TCIPA с помощью 2CP.
Помимо активации известных сигнальных белков Syk, SLP76 и PLCγ2, PDPN и родоцитин индуцируют фосфорилирование Akt1 (S473), PKCμ (S748), p38 (Y180 / Y182) и cPLA 2 (S505) но подавляют фосфорилирование PDK1 (S241). 2CP селективно изменяет статус фосфорилирования этих протеинкиназ, индуцированный PDPN, в сочетании с селективным ингибированием индуцированной PDPN агрегации тромбоцитов.Эти находки согласуются с предыдущими исследованиями различных сигнальных путей, предполагающими, что Akt, PKCμ, p38 и cPLA 2 являются эффекторами, расположенными ниже по течению Syk [56-59]. Фосфорилирование p38 наблюдалось при активации CLEC-2 моноцитов / макрофагов, запускаемой родоцитином [60]. BCR-опосредованная активация p38 является Lyn- и Syk-зависимой [57]. Syk также регулирует индуцированное FcγRIIA фосфорилирование LAT и активацию PI3K / Akt и p38 [61]. PKCμ соосаждение как с Syk, так и с PLC-γ1 / 2 [59], активируется в линии B-клеток DT40 Syk-зависимым образом [57].Активация cPLA 2 β-глюканом зависит от активации Syk в макрофагах [62]. Фосфорилирование PDK1 (S241) подавляется во время индуцированной PDPN агрегации тромбоцитов, которая может быть обращена 2CP. Аутофосфорилирование по Ser241 важно для активности киназы PDK1, которая негативно регулируется связыванием с 14–3-3 через сайт аутофосфорилирования PDK1 Ser241 [63]. Хотя передача сигналов PI3K / PDK1 / Akt является нижестоящими эффекторами Syk [58, 61, 64] и связана с активацией и агрегацией тромбоцитов [65, 66], фосфорилирование по Ser241 и связывание с 14–3-3 наиболее вероятно участвует в контроле PDPN-индуцированной активности PDK1.Эти открытия закладывают основу для открытия новых сигнальных белков при активации CLEC-2.
В заключение, было синтезировано новое нецитотоксическое низкомолекулярное соединение 2CP, которое связывается с CLEC-2 и ингибирует PDPN-индуцированную активацию CLEC-2, агрегацию тромбоцитов и TCIPA. Новое терапевтическое использование 2CP и идентификация заметных сигнальных молекул CLEC-2 были обнаружены путем анализа действия 2CP на метастазы у экспериментальных животных и на активацию тромбоцитов, индуцированную PDPN.2CP имеет потенциал в качестве антиметастатического агента при раке и в качестве инструмента для выяснения молекулярного механизма PDPN-индуцированной активации CLEC-2.
МАТЕРИАЛЫ И МЕТОДЫ
Реагенты и антитела
Тромбин и АДФ были приобретены у Calbiochem (Дармштадт, Германия). Коллаген был приобретен у Chrono-Log Co. (Хавертаун, Пенсильвания). Рекомбинантный человеческий PDPN был приобретен в Sino Biological Inc. (Пекин, Китай). Родоцитин (агретин) был очищен из яда Calloselasma rhodostoma , как описано ранее [60].U46619 был приобретен у Cayman Chemical (Анн-Арбор, Мичиган). Апираза, простациклин (PGI 2 ) и бычий сывороточный альбумин (BSA) были приобретены у Sigma (Сент-Луис, Миссури). Флуоресцентные кальцеиновые красители (зеленый кальцеин-AM и красный / оранжевый кальцеин-AM) были приобретены в Invitrogen (Карлсбад, Калифорния). Антитело против PDPN человека, конъюгированное с Alexa Fluor 488, было приобретено у BioLegend Inc. (Сан-Диего, Калифорния). Гепарин (нефракционированный) любезно предоставил доктор Сян-Руэй Ляо (Университет Чанг Гун, Тайвань).НМГ (эноксапарин, клексан) получали от Sanofi Pharma Inc. (Maisons-Alfort Cedex, Франция). CDDP был любезно предоставлен доктором Джо-Чи Ценг (Chang Gung Memory Hospital, Тайвань). Моноклональное антитело против фосфотирозина (4G10) было приобретено в EMD Millpore (Billerica, MA). Антифосфоспецифические антитела к PLCγ2 (Y1217), SLP-76 (Y145), Syk (Y525 / 526), Akt1 (S473), PDK1 (S241), PKCμ (S748), cPLA 2 (S505) и p38 (Y180 / 182) были приобретены у Cell Signaling Technology Inc. (Беверли, Массачусетс).Моноклональные антитела против PDPN крысы и антитела против β-актина были приобретены в Novus Biological (Милл-Вэлли, Калифорния). Набор для анализа активности LDH был приобретен у Biochain Inc. (Хейворд, Калифорния). Набор для анализа активности каспаз (набор Caspase-Glo 3/7) был приобретен у Promega Inc. (Мэдисон, Висконсин)
Животные, клеточные линии и поставка соединения
Все животные и протоколы экспериментов были одобрены Институтом по уходу и использованию животных. Комитет (IACUC) Университета Чанг Гунг (Тайвань, Китайская Республика) с идентификаторами утверждения CGU10–004 и CGU12–057.Голых мышей BALB / c покупали в Национальном центре лабораторных животных (Тайбэй, Тайвань) и содержали в условиях, свободных от патогенов. Клеточная линия глиомы крысы (C6, BCRC-860046) и клеточные линии остеосаркомы человека (MG63, BCRC-60279; HOS, BCRC-60308) были приобретены в Центре сбора и исследования биоресурсов (BCRC, Тайвань). C6 / LG был получен из клеток глиобластомы C6 крысы путем экспрессии слитых репортерных генов люциферазы и зеленого флуоресцентного белка (LG). C6 / Lung представляет собой подлинию C6 / LG, полученную путем восстановления метастатических колоний в легких, которые образовались при внутривенном введении клеток C6 / LG мышам nude.Все исследуемые низкомолекулярные соединения были синтезированы, как описано в дополнительных методах.
Забор крови человека
Все экспериментальные протоколы и процедуры были одобрены Советом по надзору учреждения Мемориальной больницы Чанг Гун (Линкоу, Тайвань, Китайская Республика) с идентификаторами одобрения 99–0404B и 101–3497B. Для этого исследования были набраны здоровые добровольцы, не имевшие в анамнезе гематологических заболеваний, тромбоцитов или нарушений свертывания крови или принимавших лекарства, которые могут влиять на гематологическую функцию, и подписали письменное информированное согласие.
Приготовление промытых тромбоцитов человека
Свежая кровь была взята у информированных здоровых добровольцев, не принимавших никаких лекарств, по крайней мере, за две недели до этого исследования. Для приготовления промытых тромбоцитов человека [67] богатую тромбоцитами плазму (PRP) получали из цельной крови, смешанной с 3,2% тринатрийцитратом (9: 1), центрифугированием при 200 g в течение 20 мин. Затем тромбоциты получали из PRP центрифугированием (900 g, 10 мин) и дважды промывали буфером Тирода (137 мМ NaCl, 2.65 мМ KCl, 12 мМ NaHCO 3 , 0,43 мМ NaH 2 PO 4 , 2 мМ CaCl 2 , 1 мМ MgCl 2 , 5 мМ глюкоза, 5 мМ HEPES, pH 7,35), содержащий 0,5 мкМ PGI 2 и 0,2 ед. / Мл апиразы. Наконец, промытые тромбоциты ресуспендировали в буфере Тирода и доводили до 3 × 10 8 / мл для агрегации тромбоцитов или 1 × 10 9 / мл для анализов агрегации тромбоцитов, индуцированной PDPN или опухолевыми клетками.
Агрегация тромбоцитов и TCIPA
Промытую суспензию тромбоцитов предварительно инкубировали с растворителем-носителем или тестируемыми соединениями в течение 3 минут при 37 ° C при перемешивании при 900 об / мин [67].Затем агрегация тромбоцитов вызывалась добавлением PDPN (4 мкг), тромбина (0,1 Ед / мл), коллагена (2 мкг / мл), U46619 (2 мкМ), ADP (20 мкМ) или опухолевых клеток (1,5 × 10 мкМ). 6 ячеек). Агрегацию тромбоцитов и TCIPA отслеживали и регистрировали в течение 15-20 минут с помощью агрегометра с программным обеспечением для обработки Aggro / Link (Chrono-Log Corp., Хавертаун, Пенсильвания). Время достижения 50% максимальной агрегации регистрировали и определяли как время агрегации.
Для наблюдения агрегации опухолевых клеток и тромбоцитов с помощью флуоресцентной микроскопии, тромбоциты или опухолевые клетки были предварительно загружены 2 мкМ флуоресцентными красителями для отслеживания клеток (зеленый кальцеин-AM для тромбоцитов и красный / оранжевый кальцеин-AM для опухолевых клеток) при 37 °. C в течение 30 мин, затем промыли, повторно суспендировали и скорректировали количество клеток для использования.Вкратце, тромбоциты, меченные зеленым кальцеином-AM (1 × 10 9 / мл, зеленые), инкубировали с клетками, меченными оранжевым / красным цветом кальцеина-AM (1,5 × 10 6 , красный), в реакционной пробирке для запуска TCIPA в агрегометре. Реакцию прекращали, когда степень агрегации клеток C6 / легких достигала 30% светопропускания. Все реакционные смеси были удалены из пробирки и помещены на предметное стекло для флуоресцентного анализа. Флуоресцентные гетероагрегаты тромбоцитов и опухолевых клеток исследовали с помощью микроскопа Zeiss Axiovert 200M (Carl Zeiss, Германия).
Анализы цитотоксичности с высвобождением лактозодегидрогеназы и измерением активности каспазы
Набор для анализа активности ЛДГ использовали для оценки цитотоксических эффектов 2CP на тромбоциты и культуральные клетки. Анализ проводили, как описано производителем. Вкратце, тромбоциты (1 × 10 9 / мл) инкубировали с 2CP (10–100 мкМ) и перемешивали при 900 об / мин в течение 15 мин. Супернатанты для измерения ЛДГ собирали первым центрифугированием при 3000 g в течение 5 минут с последующим центрифугированием при 10000 g в течение 5 минут.Для опухолевых клеток (1 × 10 6 ) монослойные конфлюэнтные клетки обрабатывали 2CP (20 мкМ) в течение 0–12 ч и супернатанты собирали центрифугированием, как описано выше. Супернатанты (100 мкл) смешивали в оптическом 96-луночном плоском микропланшете с 45 мкл реакционной смеси для анализа, содержащей диафоразу, NAD + , тетразолий INT и лактат. После инкубации при комнатной температуре в течение 30 мин реакцию останавливали добавлением 50 мкл стоп-раствора. Активность ЛДГ образца определяли путем измерения оптической плотности на длине волны 490 нм.В качестве контроля супернатант тромбоцитов / клеток, лизированных 1% тритоном Х-100, использовали в качестве контроля для измерения общего количества ЛДГ.
Измерение активности каспазы выполняли с использованием набора для анализа Caspase-Glo 3/7 в соответствии с инструкциями производителя. Вкратце, тромбоциты (1 × 10 8 ) или опухолевые клетки (4 × 10 3 ) помещали в 96-луночный белый микропланшет в отсутствие или в присутствии 2CP (20 или 100 мкМ) в различные моменты времени (0 , 0,25, 4 и 24 ч) при 37 ° С.Затем к клеткам добавляли равный объем реагента для анализа Caspase-Glo. Реакционные смеси инкубировали 1 ч при 37 ° C. Сигнал люминесценции измеряли с помощью люминометра SpectraMax (Molecular Devices). Клетки, обработанные стауроспорином (2 мкМ), использовали в качестве положительного контроля активности каспазы.
Экспериментальные анализы метастазов в легкие, потери массы тела и выживаемости животных
Раковые клетки (1,75 × 10 5 /100 мкл) внутривенно вводили мышам BALB / c-nu / nu в возрасте 7–8 недель.Тестируемые соединения вводили мышам внутривенно (2CP, 3,5 мг / кг; и CDDP, 2,5 мг / кг). На 15 день после инъекции опухолевых клеток с использованием системы флуоресцентного изображения Xenogen IVIS-100 (Xenogen, Alameda, CA) измеряли in vivo, биолюминесцентный анализ изображений метастатических очагов легких. Относительную область интенсивности (R.O.I) анализировали на рост опухоли. Ткань легкого была удалена и окрашена гематоксилином и эозином для гистологического исследования под микроскопом Zeiss PrimoStar (Carl Zeiss, Геттинген, Германия).Метастатические области легких анализировали и количественно оценивали с помощью программного обеспечения Zeiss AxioVision (Carl Zeiss, Германия). Соответствующая метастатическая область выражалась в процентах от всего легкого. Потерю массы тела регистрировали на 21 день после инокуляции опухоли. Процент разницы в весе рассчитывали с использованием уравнения [(День 0 — День 21) / День 0] x 100%. Выживаемость животных регистрировалась, и данные анализировались с использованием кривой выживаемости Каплана-Мейера и критерия логарифмического ранга.
Анализ времени кровотечения из хвоста
Анализ времени кровотечения из хвоста выполняли, как описано ранее [67–68]. Вкратце, мышам (7-8 недель) внутривенно вводили 2CP (3,5 мг / кг), гепарин (2 мг / кг, что эквивалентно 300 МЕ / кг), НМГ (2 мг / кг, что эквивалентно 200 МЕ / кг). ) или управление транспортным средством. Через десять минут после инъекции мышей анестезировали и 0,5 см дистального конца хвоста вырезали скальпелем. Ампутированный хвост немедленно погружали в физиологический раствор, предварительно нагретый до 37 ° C.Время кровотечения регистрировали с момента рассечения до прекращения кровотока. Наблюдения прекращались на 360 с, даже если кровотечение не прекращалось. Время кровотечения более 360 секунд было установлено как 360 секунд.
Вестерн-блоттинг
Для выделения лизатов тромбоцитов тромбоциты смешивали с 5-кратным ледяным буфером для лизиса (100 мМ Трис-HCl, pH 7,5, 300 мМ NaCl, 5% Тритон X-100, 5 мМ EGTA, 5 мМ ЭДТА, 5 мМ ФМСФ, 5 мМ Na 3 VO 4 , 20 мкг / мл лейпептина и 20 мкг / мл апротинина) и выдерживали на льду в течение 4 ч [67].Белки солюбилизировали добавлением буфера для образца до конечной концентрации 60 мМ Трис-HCl pH 6,8, 2% SDS, 10% глицерина, 0,1% бромфенолового синего и 50 мМ DTT. Затем лизаты кипятили при 95 ° C в течение 10 минут с последующим электрофорезом в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE). Для выделения клеточных лизатов культивированные клетки (2 × 10 6 / мл) собирали и дважды промывали ледяным 1X PBS. Затем клетки солюбилизировали в 1X ледяном буфере для лизиса [69] и держали на льду в течение 4 часов.После первого центрифугирования при 3000 g в течение 10 минут для удаления клеточного дебриса супернатанты собирали для второго центрифугирования (10000 g, 5 минут) с последующим количественным определением белка.
Экстрагированные белки фракционировали с помощью SDS-PAGE и переносили на PVDF-мембрану (Pall Corp., Ann Arbor, MI) с использованием системы электропереноса Bio-Rad. Мембраны блокировали 5% обезжиренным сухим молоком при 4 ° C в течение ночи и инкубировали с указанным первичным антителом, которое распознает желаемые мишени.После четырехкратной промывки трис-буферным физиологическим раствором, содержащим 0,1% Твин 20 (TBS-T), мембраны инкубировали с конъюгированным с HRP вторичным антителом (1: 20000) в течение 45 минут при комнатной температуре. После двукратной промывки TBS-T белки детектировали с использованием набора для детектирования усиленной хемилюминесценции (Millipore Corp., Бедфорд, Массачусетс).
Молекулярный стыковочный модуль соединения-кандидата
Для исследований стыковки трехмерная структура 2CP была построена с использованием Chem 3D ultra 9.0 программное обеспечение Стандарт чертежа химической структуры (CambridgeSoft Corporation, Уолтем, Массачусетс). Кристаллическая структура CLEC-2 (PDB ID 3WSR) была получена из банка данных белков RCSB. Молекулы воды удаляли, а атомы водорода добавляли к белку с помощью Discovery Studio 4.0 (Accelrys, Сан-Диего, Калифорния). Функцию силового поля CHARMm использовали для приблизительного поиска конформаций, когда соединения стыковались с CLEC-2, а затем модифицированный активный сайт CLEC-2 был выбран в качестве сайта связывания для исследования.Конформации 2CP были оптимизированы с использованием той же функции силового поля, а затем гибко состыкованы пошаговым образом с протоколом док-лигандов (Libdock) в Discovery Studio 4.0 и оценками LibDock как оценочными функциями.
Анализ связывания с поверхностным плазмонным резонансом
Аффинность связывания 2CP с CLEC-2 измеряли с помощью прибора Biacore SPR Biacore T200 (GE Healthcare, Упсала, Швеция). Количество связанного соединения с белком CLEC-2 выражалось в единицах ответа биосенсора (RU).Все анализы проводили при 25 ° C в рабочем буфере PBS-P / 1% DMSO (0,005% поверхностно-активного вещества P20 и 1% DMSO в буфере PBS). Неспецифическое связывание устраняли и контролировали путем вычитания сигнала эталонной проточной кюветы и промывки буфером. Значения и сенсограммы K D анализировали с помощью программного обеспечения Biacore Evaluation Software 1.0 (GE Healthcare). Для экспериментов по кинетическому связыванию рекомбинантный CLEC-2 (20 мкг / мл) был иммобилизован на поверхности биосенсорного чипа CM5 путем связывания с амином (10 мМ ацетат, pH 5.0) согласно протоколу производителя. Затем различные концентрации (от 2,5 до 60 мкМ) 2CP вводили в течение 2 минут при скорости потока 30 мкл / мл на предварительно иммобилизованную поверхность чипа CLEC-2 для регистрации взаимодействия 2CP-CLEC-2 с последующим расчетом константы равновесной диссоциации. (K D ).
Статистический анализ
Все эксперименты повторяли не менее трех раз, и результаты выражали в виде средних значений ± стандартная ошибка. Различия между группой, обработанной образцом, и контрольной группой анализировали с помощью непарного теста Стьюдента t , теста Манна-Уитни U и однофакторного дисперсионного анализа с использованием статистического программного обеспечения Prism версии 4.0 (Сан-Диего, Калифорния, США), где это необходимо. Выживаемость животных оценивали по кривой Каплана-Мейера и логарифмическому ранговому критерию. Результаты считались статистически значимыми при , при значении P <0,05.
ПОДТВЕРЖДЕНИЕ
Мы хотели бы поблагодарить д-ра Арнольда Стерна (Медицинский факультет Нью-Йоркского университета) за редакционную помощь и технические советы г-на Фореста Ли (GE Healthcare, Тайвань) и д-ра Шэн-Вей Линя (IBS). , Национальный университет Тайваня) за отличную техническую помощь в анализе SPR.
КОНФЛИКТ ИНТЕРЕСОВ
Авторы заявляют об отсутствии конфликта интересов.
ПОДДЕРЖКА ГРАНТА
Эта работа была частично поддержана грантами Министерства науки и технологий (NSC99–2632-B-182–001-MY3, MOST102–2628-B-182–009-MY3 и MOST102–2628- B-182–010-MY3 — C.-P. Tseng и MOST102–2320-B-182–008-MY3 — P.-W. Hsieh), а также гранты Мемориальной больницы Chang Gung (CMRPD1C0551–3, CMRPD1B0391– 3 и CMRPD1E0181), а также грант Исследовательского центра молекулярной медицины Чанг Гун (EMRPD1E1491) C.-П. Ценг.
СокращенияBMNS, 4-O-бензоил-3-метоксибета-нитростирол; CDDP, цисплатин; CLEC-2, лектин-подобный рецептор 2 С-типа; цитозольная фосфолипаза A2: cPLA2; ЛДГ, лактозодегидрогеназа; НМГ, низкомолекулярный гепарин; PDK1, киназа 1 пируватдегидрогеназы; ПДПН, подпланин; PGI2, простациклин I2; PKCμ, протеинкиназа C mu; PLCγ2, фосфолипаза C гамма 2; PRP, плазма, обогащенная тромбоцитами; SDS-PAGE, электрофорез в полиакриламидном геле с додецилсульфатом натрия; ППР — поверхностный плазмонный резонанс; TCIPA, агрегация тромбоцитов, индуцированная опухолевыми клетками.
СПИСОК ЛИТЕРАТУРЫ
1. Гупта Г.П., Массаг Дж. Повторное обращение к тромбоцитам и метастазам: новая жировая связь. J Clin Invest. 2004; 114: 1691–1693.
2. Stenger D, Dutting S, Nieswandt B. Механическое объяснение вклада тромбоцитов в метастазирование рака. Thromb Res. 2014; 133: S149–157.
3. Юрас П., Стюарт М.В., Радомски А., Хадур Ф., Душик М., Радомски М.В. Роль фактора фон Виллебранда в агрегации тромбоцитов, индуцированной опухолевыми клетками: дифференциальная регуляция NO и простациклином.Br J Pharmacol. 2001; 134: 1104–1112.
4. Jurasz P, Alonso-Escolano D, Radomski MW. Взаимодействие тромбоцитов с раком: механизмы и фармакология агрегации тромбоцитов, индуцированной опухолевыми клетками. Br J Pharmacol. 2004; 143: 819–826.
5. Эрпенбек Л., Шон М.П. Смертельные союзники: фатальное взаимодействие тромбоцитов и метастазирующих раковых клеток. Кровь. 2010; 115: 3427–3436.
6. Lian L, Li W, Li ZY, Mao YX, Zhang YT, Zhao YM, Chen K, Duan WM, Tao M. Ингибирование агрегации тромбоцитов, вызванной раком груди MCF-7, с использованием комбинации антитромбоцитарных препаратов.Oncol Lett. 2013; 5: 675–680.
7. Алонсо-Эсколано Д., Стронгин А.Ю., Чунг А.В., Дерюгина Е.И., Радомский М.В. Мембранная матричная металлопротеиназа типа 1 стимулирует агрегацию тромбоцитов, индуцированную опухолевыми клетками: роль рецепторных гликопротеинов. Br J Pharmacol. 2004; 141: 241–252.
8. Медина С., Хармон С., Инкилевич И., Сантос-Мартинес М. Дж., Джонс М., Кантвелл П., Базу Д., Ледвидж М., Радомски М. В., Гилмер Дж. Ф.. Дифференциальное ингибирование индуцированной опухолевыми клетками агрегации тромбоцитов никотинатным пролекарством аспирина (ST0702) и аспирином.Br J Pharmacol. 2012; 166: 938–949.
9. Коэн С.А., Триха М, Машелли М.А. Возможные будущие клинические применения антагониста GPIIb / IIIa, абциксимаба при тромбозах, сосудистых и онкологических показаниях. Pathol Oncol Res. 2000; 6: 163–174.
10. Амирхосрави А., Муса С.А., Амая М., Блайдес С., Десаи Х., Мейер Т., Фрэнсис Дж.Л. Ингибирование индуцированной опухолевыми клетками агрегации тромбоцитов и метастазов в легких пероральным антагонистом GpIIb / IIIa XV454. Thromb Haemost. 2003; 90: 549–554.
11.Радомский А., Юрас П., Сандерс Э. Дж., СМ в целом, Бигг Х.Ф., Эдвардс Д.Р., Радомски М.В. Идентификация, регуляция и роль тканевого ингибитора металлопротеиназы-4 (ТИМП-4) в тромбоцитах человека. Br J Pharmacol. 2002; 137: 1330–1338.
12. Клерк С.П., Сморенбург С.М., Оттен Х.М., Ленсинг А.В., Принс М.Х., Пиовелла Ф., Прандони П., Бос М.М., Ришель Д.Д., ван Тиенховен Г., Буллер HR. Влияние низкомолекулярного гепарина на выживаемость у пациентов с запущенными злокачественными новообразованиями. J Clin Oncol. 2005; 23: 2130–2135.
13.Lian L, Li W, Li ZY, Mao YX, Zhang YT, Zhao YM, Chen K, Duan WM, Tao M. Ингибирование агрегации тромбоцитов, вызванной раком молочной железы MCF-7, с использованием комбинации антитромбоцитарных препаратов. Oncol Lett. 2013; 5: 675–680.
14. Xu Y, Ogose A, Kawashima H, Hotta T., Ariizumi T., Li G, Umezu H, Endo N. Высокий уровень экспрессии подпланина в доброкачественных и злокачественных опухолях мягких тканей: иммуногистохимическая и количественная RT- ПЦР-анализ. Oncol Rep.2011; 25: 599–607.
15. Astarita JL, Acton SE, Turley SJ.Подопланин: новые функции в развитии, иммунной системе и раке. Фронт Иммунол. 2012; 3: 283.
16. Кунита А., Кашима Т.Г., Моришита Ю., Фукаяма М., Като Ю., Цуруо Т., Фудзита Н. Фактор аггруса / подопланин, вызывающий агрегацию тромбоцитов, способствует метастазированию в легкие. Am J Pathol. 2007; 170: 1337–1347.
17. Suzuki-Inoue K, Kato Y, Inoue O, Kaneko MK, Mishima K, Yatomi Y, Yamazaki Y, Narimatsu H, Ozaki Y. Участие рецептора змеиного токсина CLEC-2 в опосредованной подпланином активации тромбоцитов. раковыми клетками.J Biol Chem. 2007; 282: 25993–26001.
18. Christou CM, Pearce AC, Watson AA, Mistry AR, Pollitt AY, Fenton-May AE, Johnson LA, Jackson DG, Watson SP, O’Callaghan CA. Почечные клетки активируют рецептор тромбоцитов CLEC-2 через подпланин. Biochem J. 2008; 411: 133–140.
19. Одзаки Ю., Сузуки-Иноуэ К., Иноуэ О. Новые взаимодействия в биологии тромбоцитов: CLEC-2 / подпланин и ламинин / GPVI. J Thromb Haemost. 2009; 7: 191–194.
20. Уотсон С.П., Герберт Дж. М., Поллитт А. Ю.. GPVI и CLEC-2 в гемостазе и сосудистой целостности.J Thromb Haemost. 2010; 8: 1456–1467.
21. Северин С., Поллитт А.Ю., Наварро-Нуньес Л., Нэш Калифорния, Моурао-Са Д., Эбл Д.А., Сенис Ю.А., Уотсон С.П. Syk-зависимое фосфорилирование CLEC-2: новый механизм передачи сигналов мотива активации на основе тирозина геммунорецептора. J Biol Chem. 2011; 286: 4107–4116.
22. Судзуки-Иноуэ К., Иноуэ О., Одзаки Ю. Новый рецептор активации тромбоцитов CLEC-2: от открытия к перспективам. J Thromb Haemost. 2011; 9: 44–55.
23. Викки А., Кристофори Г.Потенциальная роль подпланина в опухолевой инвазии. Br J Рак. 2007; 96: 1–5.
24. Райка М., Симпеан А.М., Рибатти Д. Роль подпланина в прогрессировании опухоли и метастазировании. Anticancer Res. 2008; 28: 2997–3006.
25. Takagi S, Sato S, Oh-hara T., Takami M, Koike S, Mishima Y, Hatake K, Fujita N. Тромбоциты способствуют росту опухоли и метастазированию через прямое взаимодействие между Aggrus / podoplanin и CLEC-2. PLoS One. 2013; 8: e73609.
26. Судзуки-Иноуэ К., Иноуэ О, Динг Джи, Нисимура С., Хокамура К., Это К., Кашиваги Х., Томияма Ю., Ятоми Ю., Умемура К., Шин И., Хирасима М., Одзаки Ю.Основные in vivo роли рецептора лектина C-типа CLEC-2: эмбриональная / неонатальная летальность мышей с дефицитом CLEC-2 из-за неправильных соединений крови / лимфатических сосудов и нарушения образования тромба тромбоцитов с дефицитом CLEC-2. J Biol Chem. 2010; 285: 24494–24507.
27. Hsieh PW, Chang YT, Chuang WY, Shih HC, Chiang SZ, Wu CC. Синтез и биологическая оценка антитромбоцитарных и цитотоксических бета-нитростиролов. Bioorg Med Chem. 2010; 18: 7621–7627.
28. Hsieh PW, Chiang SZ, Wu CC, Lo YC, Shih YT, Wu YC.Синтез и оценка антитромбоцитов аналогов 2-бензоиламинобензоата. Bioorg Med Chem. 2008; 16: 5803–5814.
29. Hsieh PW, Hwang TL, Wu CC, Chiang SZ, Wu CI, Wu YC. Оценка и взаимосвязь структура-активность эфиров 2-бензоиламинобензойной кислоты и их аналогов в качестве противовоспалительных агентов и агентов, препятствующих агрегации тромбоцитов. Bioorg Med Chem Lett. 2007; 17: 1812–1817.
30. Wang WY, Hsieh PW, Wu YC, Wu CC. Синтез и фармакологическая оценка новых производных бета-нитростирола как ингибиторов тирозинкиназы с сильной антиагрегантной активностью.Biochem Pharmacol. 2007; 74: 601–611.
31. Wang WY, Wu YC, Wu CC. Предотвращение активации гликопротеина IIb / IIIa тромбоцитов с помощью 3,4-метилендиокси-бета-нитростирола, нового ингибитора тирозинкиназы. Mol Pharmacol. 2006; 70: 1380–1389.
32. Амирхосрави А., Муса С.А., Амая М., Фрэнсис Дж.Л. Антиметастатический эффект низкомолекулярного гепарина тинзапарина. J Thromb Haemost. 2003; 1: 1972–1976.
33. Муса С.А., Линхардт Р., Фрэнсис Дж. Л., Амирхосрави А. Антиметастатический эффект неантикоагулянтного низкомолекулярного гепарина по сравнению со стандартным низкомолекулярным гепарином, эноксапарином.Thromb Haemost. 2006; 96: 816–821.
34. Stocking KL, Jones JC, Everds NE, Buetow BS, Roudier MP, Miller RE. Использование низкомолекулярного гепарина для снижения смертности мышей после внутрисердечной инъекции опухолевых клеток. Comp Med. 2009; 59: 37–45.
35. Трэверс Р. Дж., Шеной Р. А., Калатоттукарен М. Т., Кижаккедату Дж. Н., Моррисси Дж. Х. Нетоксичные ингибиторы полифосфатов уменьшают тромбоз, сохраняя гемостаз. Кровь. 2014; 124: 3183–3190.
36. Bastida E, Ordinas A. Вклад тромбоцитов в образование метастатических очагов: роль активации тромбоцитов, индуцированной раковыми клетками.Гемостаз. 1988; 18: 29–36.
37. Цуруо Т., Фудзита Н. Агрегация тромбоцитов в образовании метастазов опухоли. Proc Jpn Acad Ser B Phys Biol Sci. 2008; 84: 189–198.
38. Fujita N, Takagi S. Влияние Aggrus / podoplanin на агрегацию тромбоцитов и метастазирование опухоли. J Biochem. 2012; 152: 407–413.
39. Nakashima Y, Yoshinaga K, Kitao H, Ando K, Kimura Y, Saeki H, Oki E, Morita M, Kakeji Y, Hirahashi M, Oda Y, Maehara Y. Подопланин экспрессируется в инвазивной передней части плоскоклеточного пищевода. клеточные карциномы и участвует в коллективной инвазии клеток.Cancer Sci. 2013; 104: 1718–1725.
40. Мартин-Виллар Э., Мегиас Д., Кастель С., Юррита М.М., Виларо С., Кинтанилла М. Подопланин связывает белки ERM, чтобы активировать RhoA и способствовать эпителиально-мезенхимальному переходу. J Cell Sci. 2006; 119: 4541–4553.
41. Cueni LN, Hegyi I, Shin JW, Albinger-Hegyi A, Gruber S, Kunstfeld R, Moch H, Detmar M. Опухолевый лимфангиогенез и метастазы в лимфатические узлы, вызванные экспрессией подопланина раковыми клетками. Am J Pathol. 2010; 177: 1004–1016.
42.Секи С., Фудзивара М., Мацуура М., Фуджита С., Икеда Х., Умеда М., Асахина И., Икеда Т. Прогностическое значение экспрессии подопланина при плоскоклеточном раке полости рта — модель регрессии, вспомогательная для классификации UICC. Pathol Oncol Res. 2014; 20: 521–528.
43. Се Дж.С., Линь Х.С., Хуанг С.Й., Хсу Х.Л., Ву Т.М., Ли К.Л., Чен М.С., Ван Х.М., Цзэн С.П. Прогностическое значение циркулирующих опухолевых клеток с экспрессией подопланина у пациентов с местнораспространенным или метастатическим плоскоклеточным раком головы и шеи. Голова Шея.2015; 37: 1448-1455.
44. Кавасе А., Исии Г., Нагаи К., Ито Т., Нагано Т., Мурата Ю., Хишида Т., Нишимура М., Йошида Дж., Судзуки К., Очиаи А. Экспрессия подопланина фибробластами, связанными с раком, предсказывает плохой прогноз аденокарциномы легких. Int J Cancer. 2008; 123: 1053–1059.
45. Накаяма Ю., Мацумото К., Нагато М., Иноуэ Ю., Кацуки Т., Минагава Н., Шибао К., Цурудоме И., Хирата К., Хигуре А., Сако Т., Нагата Н. Значимость лимфангиогенеза по оценке иммуногистохимии в пациенты с карциномой пищевода.Anticancer Res. 2007; 27: 619–625.
46. Чао Ю.К., Чуанг В.Й., Йе СиДжей, Ву Ю.К., Лю Ю.Х., Се М.Дж., Ченг А.Дж., Сюэ К., Лю Х.П. Прогностическое значение высокой экспрессии подпланина после химиолучевой терапии у пациентов с плоскоклеточным раком пищевода. J Surg Oncol. 2012; 105: 183–188.
47. Като Ю., Канеко М.К., Куно А., Учияма Н., Амано К., Чиба И., Хасегава Ю., Хирабаяси Дж., Наримацу Н., Мисима К., Осава М. Ингибирование агрегации тромбоцитов, индуцированной опухолевыми клетками, с использованием нового анти- антитело к подопланину, реагирующее со своим доменом, стимулирующим агрегацию тромбоцитов.Biochem Biophys Res Commun. 2006; 349: 1301–1307.
48. Канеко М.К., Кунита А., Абэ С., Цудзимото Ю., Фукаяма М., Гото К., Сава Ю., Нисиока Ю., Като Ю. Химерное антитело против подопланина подавляет метастазирование опухоли посредством нейтрализации и антителозависимой клеточной цитотоксичности. Cancer Sci. 2012; 103: 1913–1919.
49. Абэ С., Морита Ю., Канеко М.К., Ханибути М., Цудзимото Ю., Гото Х., Какиучи С., Аоно Ю., Хуанг Дж., Сато С., Кишуку М., Танигучи Ю., Адзума М. и др. Новая нацеленная терапия злокачественной мезотелиомы с использованием антител против подопланина.J Immunol. 2013; 190: 6239–6249.
50. Накадзава Ю., Такаги С., Сато С., Ох-хара Т., Коике С., Таками М., Араи Х, Фудзита Н. Профилактика гематогенных метастазов путем нейтрализации мышей и их химерных антител против агруса / подопланина. Cancer Sci. 2011; 102: 2051–2057.
51. Като Ю., Вайдьянатан Г., Канеко М.К., Мисима К., Шривастава Н., Чандрамохан В., Пеграм С., Кейр С.Т., Куан С.Т., Бигнер Д.Д., Залуцкий М.Р. Оценка крысиного моноклонального антитела против подопланина NZ-1 для нацеливания на злокачественные глиомы.Nucl Med Biol. 2010; 37: 785–794.
52. Чандрамохан В., Бао Х, Като Канеко М., Като Ю., Кейр С.Т., Шафрански С.Е., Куан С.Т., Пастан И.Х., Бигнер Д.Д. Рекомбинантный иммунотоксин против подопланина (NZ-1) для лечения злокачественных опухолей головного мозга. Int J Cancer. 2013; 132: 2339–2348.
53. Пандей А., Саранги С., Чиен К., Сенгупта П., Папа А.Л., Басу С., Сенгупта С. Антитромбоцитарные средства повышают цитотоксичность наночастиц цисплатина за счет увеличения проницаемости сосудистой сети опухоли и доставки лекарств. Нанотехнологии.2014; 25: 445101.
54. Nagae M, Morita-Matsumoto K, Kato M, Kaneko MK, Kato Y, Yamaguchi Y. Платформа лектин-подобного рецептора C-типа CLEC-2 для связывания O-гликозилированного подопланина и негликозилированного родоцитина. Состав. 2014; 22: 1711–1721.
55. Уотсон А.А., Браун Дж., Харлос К., Эбл Дж. А., Уолтер Т.С., О’Каллаган, Калифорния. Кристаллическая структура и анализ мутационного связывания внеклеточного домена активирующего тромбоциты рецептора CLEC-2. J Biol Chem. 2007; 282: 3165–3172.
56.Jackson EC, Ortar G, McNicol A. Влияние ингибитора диглицерид липазы на активацию тромбоцитов, индуцированную коллагеном. J Pharmacol Exp Ther. 2013; 347: 582–588.
57. Цзян А., Кракстон А., Куросаки Т., Кларк Э.А. Различные протеинтирозинкиназы необходимы для опосредованной рецептором В-клетки активации внеклеточной киназы, регулируемой сигналом, Nh3-концевой киназы 1 c-Jun и протеинкиназы, активируемой митогеном p38. J Exp Med. 1998; 188: 1297–1306.
58. Лау К., Ван Х, Сонг Л., Норт М., Вилер С., Гордый Д., Чоу К. В..Syk связывается с клатрином и опосредует активацию фосфатидилинозитол-3-киназы во время интернализации человеческого риновируса. J Immunol. 2008; 180: 870–880.
59. Сидоренко С.П., Юриспруденция К.Л., Клаус С.Дж., Чандран К.А., Таката М., Куросаки Т., Кларк Е.А. Протеинкиназа C mu (PKC mu) связывается с рецепторным комплексом B-клеточного антигена и регулирует передачу сигналов лимфоцитами. Иммунитет. 1996; 5: 353–363.
60. Чанг СН, Чжун СН, Сюй СС, Пэн ХК, Хуанг Т.Ф. Ингибирующее действие полипептидов, полученных из лектина С-типа змеиного яда, агретина, на индуцированную опухолевыми клетками агрегацию тромбоцитов.J Thromb Haemost. 2014; 12: 540–549.
61. Lhermusier T, van Rottem J, Garcia C, Xuereb JM, Ragab A, Martin V, Gratacap MP, Sie P, Payrastre B. Ингибитор Syk-киназы R406 нарушает активацию тромбоцитов и экспрессию тканевого фактора моноцитов, запускаемую гепарином. Комплексные антитела PF4. J Thromb Haemost. 2011; 9: 2067–2076.
62. Сурам С., Браун Г.Д., Гош М., Гордон С., Лопер Р., Тейлор П.Р., Акира С., Уэмацу С., Уильямс Д.Л., Лесли С.К. Регулирование активации цитозольной фосфолипазы А2 и экспрессии циклооксигеназы 2 в макрофагах с помощью рецептора бета-глюкана.J Biol Chem. 2006; 281: 5506–5514.
63. Сато С., Фудзита Н., Цуруо Т. Регулирование киназной активности 3-фосфоинозитид-зависимой протеинкиназы-1 путем связывания с 14–3-3. J Biol Chem. 2002; 277: 39360–39367.
64. Hemmings BA, Restuccia DF. Путь PI3K-PKB / Akt. Cold Spring Harb Perspect Biol. 2012; 4: a011189.
65. Woulfe DS. Передача сигналов Akt в тромбоцитах и тромбозе. Эксперт Рев Гематол. 2010; 3: 81–91.
66. Чен Х, Чжан И, Ван И, Ли Д, Чжан Л., Ван К., Ло Х, Ян З, Ву И, Лю Дж.PDK1 регулирует активацию тромбоцитов и артериальный тромбоз. Кровь. 2013; 121: 3718–3726.
67. Цай Х.Дж., Хуанг К.Л., Чанг Ю.В., Хуанг Д.Й., Лин С.К., Купер Д.А., Ченг Дж.С., Цзэн С.П. Disabled-2 необходим для эффективного гемостаза и активации тромбоцитов тромбином у мышей. Артериосклер Thromb Vasc Biol. 2014; 34: 2404–2412.
68. Ценг В.Л., Чен Т.Х., Хуанг С.К., Хуанг Й.Х., Йе К.Ф., Таси Х.Д., Ли Х.Й., Као С.Й., Лин С.В., Ляо Х.Р., Ченг Дж.С., Цзэн С.П. Нарушение генерации тромбина у Reelin-дефицитных мышей: потенциальная роль Reelin плазмы в гемостазе.J Thromb Haemost. 2014; 12: 2054–2064.
69. Цзэн С.П., Хуанг С.Ч., Цзэн С.К., Линь М.Х., Се Дж.Т., Цзэн С.П. Индукция гена disabled-2 во время дифференцировки мегакариоцитов клеток K562. Biochem Biophys Res Commun. 2001; 285: 129–135.
Границы | Амилин и кальцитонин: потенциальные терапевтические стратегии для снижения массы тела и жира в печени
Введение
Стеатоз печени широко считается печеночным проявлением метаболического синдрома (1). Параллельно с увеличением распространенности ожирения и связанных с ним заболеваний неалкогольная жировая болезнь печени (НАЖБП) в настоящее время является наиболее распространенным заболеванием печени в мире (2–4).Снижение массы тела в настоящее время является наиболее эффективной стратегией для улучшения показателей стеатоза и НАЖБП (5), и некоторые лекарства от ожирения, находящиеся в клинической разработке, продемонстрировали улучшения в отношении содержания жира в печени (6, 7). Недавние доклинические исследования продемонстрировали потерю массы тела, снижение стеатоза печени и улучшение метаболизма у крыс после введения новых двойных агонистов рецепторов амилина и кальцитонина (DACRA) (8-13).
НАЖБП определяется повышенным содержанием жира в печени (> 5%) без значительного употребления алкоголя или стеатоза, вызванного каким-либо другим механизмом (например,д., лекарства, гепатит, аутоиммунные или наследственные заболевания) (14, 15). НАЖБП охватывает спектр стадий, от простого накопления жира в печени до неалкогольного стеатогепатита (НАСГ) с воспалением и, в конечном итоге, до фиброза и цирроза печени (1). Недавно термин «метаболически-ассоциированная жировая болезнь печени (MASH)» был предложен как объединяющее определение стеатоза печени у лиц с избыточным весом / ожирением, нарушением метаболизма и / или манифестным диабетом 2 типа (16).Это определение признает важность ожирения и инсулинорезистентности, а не отсутствия чрезмерного употребления алкоголя, как причинного фактора для развития стеатоза печени. В связи с этим распространенность НАЖБП резко возрастает с увеличением количества критериев метаболического синдрома в популяции (17). Кроме того, хорошо установленная связь между НАЖБП и диабетом 2 типа подчеркивает потенциальную случайную связь между нарушением метаболизма глюкозы с инсулинорезистентностью и стеатозом печени (18).Даже при предиабете повышенное содержание жира в печени является центральной характеристикой (19). Накопление липидов в печени может способствовать развитию устойчивости как к инсулину (20), так и к глюкагону (21), которые являются важными патофизиологическими характеристиками диабета 2 типа (22, 23). Инсулинорезистентность считается движущей силой стеатоза печени из-за повышенного липогенеза печени и чрезмерного липолиза тканей, что в конечном итоге увеличивает накопление жирных кислот в печени (24).
В настоящее время бариатрическая хирургия является наиболее эффективной терапией для снижения веса, но она дорогостоящая, сопряжена с немаловажным риском осложнений, и не все пациенты подходят для операции (25).Таким образом, фармакотерапия для снижения массы тела активно проводится, и амилин, а также DACRA появляются в качестве потенциальных новых кандидатов в лекарства от ожирения, особенно в сочетании с другими снижающими массу тела пептидами желудочно-кишечного тракта.
Этот обзор суммирует физиологию и патофизиологию амилина и кальцитонина при ожирении и НАЖБП и дает представление о потенциальной терапевтической роли фармакологических доз амилина и кальцитонина, имеющих отношение к метаболическим заболеваниям, включая ожирение и НАЖБП.
Амилин
Амилин представляет собой пептидный гормон из 37 аминокислот (таблица 1), который в основном вырабатывается бета-клетками поджелудочной железы и совместно секретируется с инсулином в ответ на поступление питательных веществ (рисунок 1) (28, 29). Гормон играет хорошо известную роль как сигнал сытости; эффект, который опосредуется через прямое действие на рецепторы амилина в определенных областях мозга, то есть в постремной области и ядре единственного тракта (30, 31). Амилин также является эффективным ингибитором опорожнения желудка, что дополнительно способствует насыщению (32–34) и может подавлять секрецию глюкагона посредством центральных механизмов (35, 36).Существует несколько изотипов рецептора амилина (37), которые все являются рецепторами, связанными с G-белком, состоящими из двух единиц; основная единица, состоящая из рецептора кальцитонина (7-трансмембранный рецептор) и одного из трех белков, модифицирующих активность рецептора (RAMP1-3) (37). Стимуляция рецепторного комплекса амилина увеличивает продукцию внутриклеточного циклического аденозинмонофосфата (38, 39). Распределение рецепторного комплекса амилина в тканях трудно описать по ряду причин: 1) рецептор кальцитонина (основная единица) имеет два подтипа, которые взаимодействуют с RAMP, 2) RAMP связаны с другими рецепторами, кроме рецепторного комплекса амилина, и 3) отсутствие селективных фармакологических инструментов и антител для нацеливания на специфические рецепторные комплексы амилина (RAMP / кальцитонин) (26).Текущие данные показывают, что несколько областей мозга, включая постремную область и гипоталамус, являются важными участками действия амилина (40, 41). В модели с нокаутом мышей отсутствие рецептора кальцитонина, в частности, в нейронах, экспрессирующих проопиомеланокортин, в гипоталамусе приводит к увеличению ожирения, непереносимости глюкозы и снижению расхода энергии (41). Кроме того, рецепторы амилина не активируются селективно одним амилином и без разбора взаимодействуют с другими гормонами аналогичной структуры (т.е., кальцитонин, пептид, связанный с геном кальцитонина, и адреномедуллин) (42). Например, было показано, что кальцитонин активирует несколько подтипов рецептора амилина (42). Аналогичным образом было показано, что амилин имеет сродство как к рецепторам кальцитонина, так и к рецепторам амилина (43). Было идентифицировано несколько антагонистов рецепторов амилина, но, как упоминалось выше, эти соединения также имеют проблемы с селективностью и не различают подтипы рецепторов амилина (26). Поэтому трудно установить важность отдельного рецептора в опосредовании эндогенного действия амилина.
Таблица 1 Аминокислотные последовательности амилина, кальцитонина и родственных пептидов.
Рисунок 1 Предполагаемые физиологические эффекты активации рецепторов амилина и кальцитонина. DACRA, двойной агонист рецепторов амилина и кальцитонина; КТ — кальцитонин; синие стрелки указывают на эффекты, связанные с амилином; зеленые стрелки указывают на эффекты, связанные с кальцитонином; оранжевые стрелки указывают на эффекты, связанные с инсулином. Элементы рисунка с smart.servier.com под CC BY 3.0.
Роль амилина в ожирении
Несколько исследований указывают на роль амилина в гормональной регуляции приема пищи и веса тела.Амилин обладает несколькими характеристиками гормона насыщения: 1) он высвобождается после приема пищи (28, 29), 2) имеет короткий период полувыведения (~ 13 мин) с быстрым началом действия (44, 45) и 3 ) он дозозависимо снижает потребление пищи при введении крысам в основном в супрафизиологических дозах (40, 45, 46). Этот эффект также проявляется в испытаниях на людях агонизма рецепторов амилина (см. Дополнительные подробности ниже). Роль эндогенного амилина как агента насыщения подтверждается наблюдением, что инъекция антагониста рецептора амилина AC187 внутривенно или непосредственно в область постремы резко увеличивает потребление пищи крысами (47, 48).В дополнение к его влиянию на чувство сытости, доклинические исследования показывают, что эндогенный амилин также обладает характеристиками сигнала ожирения (т. Е. Гормонального фактора, регулирующего массу тела, циркулирующего пропорционально массе жира тела), во многом как инсулин и лептин, а именно способность для увеличения расхода энергии и снижения массы тела с помощью центральных механизмов (49, 50). У крыс хроническое внутривенное введение амилина приводит к потере массы тела и уменьшению отложения жира, тогда как централизованное введение амилина, помимо снижения массы тела, по-видимому, также снижает целевую массу тела, установленную мозгом (49–52).Подтверждая его роль в качестве сигнала эндогенного ожирения, острый и хронический антагонизм амилина с AC187 имеет противоположный эффект и увеличивает потребление пищи и массу тела крыс (50). Как сигнал ожирения амилин усиливает насыщающий эффект холецистокинина (ХЦК). Об этом свидетельствует синергетический острый эффект совместного введения CCK и амилина на потребление пищи при внутрибрюшинном введении мышам (53), а также снижение действия CCK на моделях дефицита амилина у мышей (54) и у крыс после инфузии антагонисты рецепторов амилина (55, 56).Хроническое внутрибрюшинное введение CCK снижает потребление пищи крысами, но мало влияет на массу тела из-за компенсаторного увеличения частоты приема пищи (57). Напротив, хроническая подкожная инфузия амилина снижает как размер еды, так и частоту приема пищи с сопутствующей потерей массы тела у крыс (46). Интересно, что амилин также взаимодействует с лептином в контроле энергетического метаболизма в ряде доклинических исследований, подтверждая его совместную роль в качестве сигнала насыщения и ожирения (58–61). У крыс с ожирением или функциональной резистентностью к лептину влияние агонизма амилина на пищевое поведение все еще наблюдается в течение 24 ч после инъекции агониста амилина (58).Кроме того, внутрижелудочковое введение лептина, по-видимому, усиливает ингибирующий эффект амилина на кратковременное питание, что свидетельствует о синергизме действия этих гормонов (59). Кроме того, амилин повторно сенсибилизирует тучных крыс к лептину при совместном введении с агонистом лептина в течение 14 дней (60). В отличие от лептина, амилин демонстрирует сохраненный аноректический ответ при исследовании крыс с ожирением и гиперамилинемией (61). Фактически, несколько исследований на грызунах предполагают хорошо сохранившуюся аноректическую реакцию на острое введение амилина при ожирении и резистентности к лептину (26, 60, 61), что делает амилин многообещающим кандидатом для фармацевтической терапии для снижения веса.Кроме того, у лиц с ожирением также наблюдается аддитивный эффект при одновременном одновременном применении амилина и агонистов лептина, что добавляет дополнительные доказательства взаимодействия между лептином и амилином (60). Химические свойства человеческого амилина предрасполагают гормон к агрегации и образованию амилоидных фибрилл, которые часто обнаруживаются в островках поджелудочной железы у людей с диабетом 2 типа и, возможно, способствуют разрушению бета-клеток (62). По этой причине вливания человеческого амилина затруднены, часто требуя высоких супрафизиологических доз, чтобы вызвать незначительный эффект или его отсутствие (63, 64).Однако было разработано несколько стабильных аналогов амилина, способных вызывать потерю массы тела (более подробная информация приводится ниже).
В совокупности амилин оказывает острое действие как сигнал сытости в сочетании с гомеостатическими эффектами сигнала ожирения на массу тела, что свидетельствует о важной роли амилина в регуляции массы тела. В клинических испытаниях уровни циркулирующего амилина тесно коррелируют с жировой массой. Исследования показывают, что базальный уровень и уровни амилина, стимулированные приемом пищи, повышены у лиц с ожирением (65–74).Это может относиться к роли амилина как регулятора массы тела, но также может быть проявлением повышенной секреторной активности бета-клеток, часто обнаруживаемой при ожирении. Эти исследования обычно ограничены размером выборки, и были опубликованы противоположные отчеты (75). Доклинические исследования подтверждают мнение о повышенном уровне амилина у крыс с ожирением (76, 77). Это может быть результатом снижения чувствительности к амилину после продолжительной гиперамилинемии, но в настоящее время доказательств этого у людей нет.Необходимы дополнительные исследования, специально предназначенные для оценки секреции и чувствительности амилина у людей с ожирением по сравнению с людьми с нормальным весом.
Кальцитонин
Кальцитонин представляет собой пептидный гормон, состоящий из 32 аминокислот (таблица 1), полученный из предшественника прокальцитонина из 116 аминокислот и секретируемый С-клетками щитовидной железы. Как упоминалось выше, кальцитонин опосредует свои эффекты через 7-трансмембранный рецептор кальцитонина и последующее увеличение внутриклеточного цАМФ (38).Из-за взаимодействия с RAMP сложно определить тканевое распределение мономерного рецептора кальцитонина, но хорошо известные органы-мишени кальцитонина включают кости и почки (78, 79). У людей секреция кальцитонина стимулируется приемом кальция (80). Кальцитонин обладает сильным гипокальциемическим эффектом за счет ингибирования остеокластов (рис. 1) (79) и стимулирования почечной экскреции кальция, предположительно путем ингибирования канальцевой реабсорбции кальция (79). С момента открытия кальцитонина в 1962 г. (81) были приложены большие усилия для описания его ингибирующего действия на остеокласты и повышенной экскреции кальция у людей, в то время как другие физиологические эффекты не были описаны.Человеческий кальцитонин часто считается рудиментарным гормоном, в основном из-за того, что гиперсекреция или дефицит кальцитонина (как это наблюдается у пациентов с медуллярным раком щитовидной железы) не связаны с аномалиями костей (82). Кроме того, более мощная форма кальцитонина, полученная из лосося, была предпочтительным выбором для лечения хронических состояний с гиперкальциемией, пока не появились лучшие антирезорбтивные препараты (например, бисфосфонаты, деносумаб и ралоксифен) (83).
Кальцитонин в метаболических заболеваниях
Трудно оценить роль эндогенного кальцитонина в метаболических заболеваниях по ряду причин: 1) только несколько исследований применяли человеческий кальцитонин у людей, 2) в настоящее время нет доступных антагонистов, которые избирательно нацелены мономерный рецептор кальцитонина (26) и 3) исследования с применением более мощного кальцитонина лосося также выявили эффекты, связанные с активностью рецептора амилина (84).Как будет показано в следующих разделах, действие кальцитонина лосося и человека напрямую не сопоставимо. В нескольких исследованиях кальцитонин млекопитающих использовался для изучения эффектов, выходящих за рамки тех, которые связаны с метаболизмом кальция и костей. Интересно, что инфузии кальцитонина как человека, так и свиньи подавляют реакцию инсулина на однократное введение глюкозы у людей (85–87). Но еще предстоит определить, имеет ли этот эффект физиологическое значение. В исследовании с участием 26 субъектов, которые в основном имели избыточный вес, но с нормальной толерантностью к глюкозе, наблюдалось повышение уровня кальцитонина в сыворотке после перорального теста на толерантность к глюкозе 75 г, которое коррелировало с уровнями инсулина, что указывает на возможную связь между инсулином и кальцитонином (88 ).Это согласуется с наблюдением, что инсулин напрямую стимулирует высвобождение кальцитонина в перфузируемой щитовидной железе свиньи (89). Кроме того, сообщалось о более высоких уровнях эндогенного кальцитонина у лиц с ожирением (90). Наконец, прокальцитонин экспрессируется в жировой ткани, и его экспрессия связана с ожирением, инсулинорезистентностью и метаболическим синдромом (91). Все вместе взятые к настоящему времени исследования дают некоторые указания на то, что у человека может существовать корреляция между кальцитонином и инсулином, но исследования, специально разработанные для подтверждения этого, оправданы.
Фармакотерапия на основе амилина и кальцитонина
Прамлинтид
Прамлинтид — аналог амилина, одобренный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), разработанный для людей с диабетом 1 типа или инсулинорезистентным диабетом 2 типа в качестве дополнительной терапии к инсулину во время еды ( 92). Прамлинтид имеет фармакологические свойства, сравнимые с человеческим амилином, но с повышенной стабильностью, что делает его пригодным для подкожного введения людям (44). Это относительно короткоживущий пептид с периодом полураспада ~ 20–45 минут у человека, поэтому его необходимо вводить с каждым приемом пищи, чтобы уменьшить скачки уровня глюкозы в плазме после приема пищи (44, 93).Вдохновленный крысиным амилином, который менее склонен к димеризации, повышенная стабильность прамлинтида по сравнению с человеческим амилином была достигнута путем введения трех аминокислотных замен (про25, 28, 29) в последовательность человеческого амилина (26) (таблица 1). В дополнение к уменьшению скачков глюкозы в плазме после приема пищи, прамлинтид продемонстрировал способность снижать массу тела в нескольких клинических испытаниях (94–112). В 6-недельном рандомизированном слепом плацебо-контролируемом многоцентровом исследовании приняли участие 60 человек с ожирением (средний индекс массы тела (ИМТ) = 35.3 кг / м ( 2 ) титровали до 180 мкг прамлинтида, вводимого подкожно перед каждым приемом пищи, чтобы проверить переносимость потери веса тела, наблюдаемой в исследованиях с прамлинтидом у лиц с диабетом (94). Через 6 недель среднее изменение массы тела по сравнению с исходным уровнем составило -2,04 кг, что соответствует потере веса примерно на 2%. Это было очень важно по сравнению с группой плацебо. В 4-месячном исследовании аналогичного дизайна большинство (88%) из 108 человек с ожирением (средний ИМТ = 37.9 кг / м ( 2 ) титровали до 240 мкг прамлинтида во время еды, вводимого подкожно три раза в день (95). В результате средняя потеря веса 3,7% была очень значительной по сравнению с группой плацебо. Чтобы оценить устойчивость этих результатов, в 12-месячном рандомизированном двойном слепом плацебо-контролируемом многоцентровом исследовании с участием 146 пациентов с ожирением (средний ИМТ ~ 37 кг / м 2 ) оценивалось влияние прамлинтида в сочетании с вмешательством в образ жизни. о длительной потере массы тела (96).В течение начального 4-месячного периода повышения дозы оценивали различные дозы (120, 240 и 360 мкг) и частоту введения (два или три раза в день) прамлинтида, после чего следовало 8-месячное продление предварительно назначенного режима лечения прамлинтидом. Через 4 месяца снижение массы тела было сопоставимо с предыдущими испытаниями прамлинтида и варьировалось от 3,8 до 6,1 кг в зависимости от дозы прамлинтида. Интересно, что через 12 месяцев потеря массы тела сохранялась при всех схемах лечения прамлинтидом, за исключением тех, которые получали 120 мкг дважды в день.Это обнадеживает, поскольку постепенное восстановление массы тела — обычное наблюдение после снижения веса, вызванного образом жизни (97, 113, 114). У лиц с ожирением, страдающих диабетом 2 типа, принимающим инсулин, прамлинтид в зависимости от дозы снижает массу тела (98–104). Эффект снижения массы тела прамлинтида также наблюдается у лиц с диабетом 1 типа (105–112).
Как указано выше, лечение прамлинтидом связано со снижением массы тела на 2–6% у нескольких категорий пациентов, включая лиц с ожирением и диабетом 2 типа, которые склонны к развитию стеатоза печени.Это делает фармакологию на основе амилина многообещающим кандидатом для лечения ожирения и НАЖБП. Однако длительное лечение прамлинтидом ограничено низкой биодоступностью и коротким периодом полувыведения препарата, что делает его менее подходящим для хронической терапии из-за высокой частоты приема (два-три раза в день) и, следовательно, высоких требований, связанных с соблюдением режима лечения. . Кроме того, монотерапия прамлинтидом оказывает лишь умеренное влияние на потерю массы тела по сравнению, например, с агонистами рецептора глюкагоноподобного пептида 1 (GLP-1) (115) или более драматическими изменениями, наблюдаемыми после бариатрической хирургии (116).Фармакотерапия, нацеленная на несколько механизмов, в настоящее время активно изучается в качестве потенциальных стратегий снижения веса (117). В доклинических и клинических условиях амилин комбинируют с несколькими другими соединениями, способными регулировать вес, чтобы вызвать положительное влияние на массу тела. К ним относятся агенты на основе пептида YY (PYY), CCK, меланокортинов, лептина и GLP-1, а также низкомолекулярные аноректики (фентермин / сибутрамин) (54, 118–123). Только комбинация GLP-1 / амилин в настоящее время разрабатывается для лечения ожирения в клинических испытаниях (124).Позже в этом разделе мы рассмотрим эффект нацеливания на несколько рецепторов в семействе рецепторов кальцитонина с помощью новых мономолекулярных двойных агонистов , но сначала мы кратко рассмотрим результаты исследований с другими агентами на основе амилина.
Давалинтид
Давалинтид представляет собой агонист рецептора амилина пептида из 32 аминокислот (таблица 1) с повышенной активностью, эффективностью и продолжительностью действия по сравнению с амилином у крыс (125). Давалинтид был разработан в связи с низкой биодоступностью и коротким периодом полувыведения, которые сделали терапию прамлинтидом при ожирении неэффективной.Пептид представляет собой химеру амилина и кальцитонина лосося и имеет 49% аминокислотной последовательности крысиного амилина и прамлинтида (26, 125). Период полувыведения давалинтида составляет 26 минут, что сопоставимо с крысиным амилином (126). Однако давалинтид снижает потребление корма крысами на срок до 23 часов по сравнению с только 6 часами у крысиного амилина (125). Кроме того, давалинтид дозозависимым образом снижает массу тела и жировую массу у крыс примерно в 2 раза большей эффективностью, чем амилин крыс (125). Увеличенная продолжительность, вероятно, объясняется медленной диссоциацией рецепторов кальцитониновой части давалинтида лосося (38, 127).Действительно, анализ связывания рецептора выявил очень ограниченную диссоциацию рецептора давалинтида в прилежащем ядре крысы (126). На данный момент эти доклинические исследования давалинтида представляют собой единственную доступную литературу по давалинтиду, и дальнейшие исследования давалинтида на людях были прекращены из-за отсутствия превосходства над прамлинтидом в отношении потери веса (128). Тем не менее, эти несколько испытаний с давалинтидом иллюстрируют, как рецепторная система кальцитонина может представлять собой мишень для лечения ожирения и связанных с ним метаболических состояний, таких как НАЖБП.
Агонисты амилина длительного действия
Амилин был модифицирован различными методами (например, путем добавления к молекуле полиэтиленгликоля (ПЭГ), гликозилирования или связывающего альбумин мотива) для увеличения периода полужизни и уменьшения частоты введения , что делает его более подходящим для хронического использования в терапии похудания (129–135). У мышей подкожное введение ПЭГилированного амилина резко снижает гликемию с пролонгированным действием по сравнению с немодифицированным амилином (129). В моделях диабета 1 типа на крысах аналог пегилированного амилина предотвращал гипергликемию, вызванную приемом пищи, и способствовал устойчивой нормогликемии до 8 часов после инъекции аналога амилина (132).Важно отметить, что исследования при острых и хронических заболеваниях показывают, что аналоги амилина длительного действия снижают массу тела и потребление энергии у крыс (136, 137). Учитывая удобство инъекции один раз в день по сравнению с несколькими инъекциями в день, агонисты амилина длительного действия привлекательны для разработки и в настоящее время рассматриваются в качестве новых кандидатов в лекарственные средства против ожирения и диабета многими фармацевтическими компаниями. Ново Нордиск в настоящее время тестирует аналог амилина пролонгированного действия, AM833, у лиц с избыточным весом и ожирением в фазе I и II клинических испытаний (138, 139).В недавно опубликованном исследовании фазы II было исследовано влияние изменений образа жизни вместе с увеличением доз (0,3, 0,6, 1,2, 2,4 и 4,5 мг) AM833 один раз в неделю на массу тела у 706 человек с ожирением / избыточной массой тела (140). Через 26 недель масса тела постепенно и дозозависимо снижалась без плато, со снижением в диапазоне от 6% до 10,8%. По сравнению с плацебо и 3 мг лираглутида один раз в день наблюдаемая потеря веса среди участников, получавших AM833, была значительно больше для всех доз AM833 по сравнению с плацебо и для 4.5 мг один раз в неделю по сравнению с лираглутидом. AM833 также оценивался для использования в комбинации с семаглутидом, аналогом GLP-1, в ходе клинических испытаний фазы I (124, 141). В общей сложности 80 участников с ожирением или избыточной массой тела получали возрастающие дозы AM833 в сочетании с 2,4 мг семаглутида один раз в неделю (141). Через 20 недель участники, получавшие самую высокую дозу AM833 с семаглутидом, потеряли в среднем 17,1% массы тела от исходного уровня (141). Также Зеландия Фарма разрабатывает аналоги амилина пролонгированного действия для лечения ожирения и диабета (142, 143).Доклинические данные на моделях крыс с диабетом и ожирением были опубликованы для соединений ZP4982 и ZP5461; обе молекулы являются мощными активаторами рецепторов кальцитонина и амилина и эффективно снижают уровень глюкозы в крови и массу тела (142, 143). По сравнению с доклиническим дозированием два раза в день с аналогом GLP-1 лираглутидом, один раз в неделю дозирование ZP4982 в почти эквимолярных дозах было лучше с точки зрения потери веса на модели крыс с ожирением (143). Фаза I клинических испытаний была проведена с аналогом амилина пролонгированного действия, разработанным компанией Зеландия Фарма в сотрудничестве с Boehringer Ingelheim в 2018 году, но сотрудничество по этому аналогу было прекращено в 2020 году (144, 145).
Кальцитонин лосося
Кальцитонин, экстрагированный из лосося, демонстрирует более длительную активацию рецепторов и связывание у людей по сравнению с кальцитонином человека (38). Он также превосходит кальцитонин млекопитающих по своим гипокальциемическим эффектам у крыс и людей (146). Интересно, что кальцитонин лосося имеет не только эффекты, связанные с метаболизмом костей. В исследованиях на людях кальцитонин лосося подавляет опорожнение желудка и высвобождение гастрина после еды, вызывая дозозависимое расслабление желчного пузыря как в постпрандиальном, так и в голодном состоянии (147, 148).У мышей и обезьян кальцитонин лосося действует аноректически и вызывает потерю веса после однократного введения (149, 150). В хронических исследованиях пероральные препараты кальцитонина лосося также снижают потребление пищи и массу тела на моделях ожирения и диабета на крысах (151, 152). Кроме того, кальцитонин лосося резко стимулирует расход энергии во время ограничения пищи у крыс (153). Кальцитонин человека и лосося имеет только 50% гомологию аминокислотной последовательности (38) (таблица 1), и исследования на грызунах с применением антагонистов рецептора амилина предполагают, что аноректический эффект кальцитонина лосося является, по крайней мере, частично результатом активации рецептора амилина (84). In vitro кальцитонин лосося демонстрирует превосходное сродство связывания с рецепторами амилина без различия рецепторов амилина и кальцитонина (154). По сравнению с кальцитонином человека кальцитонин лосося также демонстрирует пролонгированную активацию рецепторов кальцитонина человека при тестировании на линиях клеток млекопитающих, экспрессирующих рецептор кальцитонина (38). Это предполагает, что кальцитонин лосося является двойным агонистом с активностью в отношении рецепторов амилина и кальцитонина , что подчеркивает возможность нацеливания на эти рецепторы с использованием одной молекулы с агонистическими свойствами двойного рецептора.
Двойные агонисты рецепторов амилина и кальцитонина
Вдохновленные фармакологией кальцитонина лосося, были разработаны DACRA для лечения ожирения и диабета (8–13). DACRA демонстрируют такое же сродство и повышенную эффективность в отношении рецепторов амилина и кальцитонина по сравнению с кальцитонином лосося (8). На крысах с диабетом Цукера с ожирением 4-недельное лечение DACRA KBP-042 от Nordic Bioscience сравнивали с кальцитонином лосося и носителем при парном вскармливании; демонстрируя значительную потерю веса и улучшенную толерантность к глюкозе по сравнению с носителем и кальцитонином лосося (8).В других исследованиях подкожные инъекции KBP-042 приводили к значительной потере массы тела с уменьшением лептиновой и инсулинорезистентности у крыс на диете с высоким содержанием жиров по сравнению с крысами на обычной диете (11, 12). Интересно, что после 7-недельного периода лечения снижение отложения жира в печени наблюдалось у крыс, получавших KBP-042, но не в контрольной группе крыс, получавших парное питание (12). Способность снижать накопление липидов в печени была также продемонстрирована с другим DACRA, KBP-089 (10). У крыс, получавших диету с высоким содержанием жиров без терапии в течение 10 недель, а затем подкожную пептидную терапию в течение 8 недель, KBP-089 полностью устранял стеатоз печени, достигнутый при начальном питании с высоким содержанием жиров (10).Важно отметить, что этот эффект не наблюдался в группе крыс, получавших парное вскармливание, что позволяет предположить, что терапия DACRA оказывает положительное влияние на стеатоз печени, помимо тех, которые связаны с уменьшением потребления пищи и потерей веса. Эти пептиды являются многообещающими с точки зрения их воздействия на массу тела и некоторые физиологические параметры, связанные с энергетическим гомеостазом и метаболизмом глюкозы в моделях ожирения и диабета на грызунах. Из доступной литературы неясно, в какой степени рецепторы амилина и кальцитонина опосредуют положительные результаты, полученные в доклинических исследованиях DACRA.В исследовании на грызунах, сравнивающем активность молекулы DACRA с природным амилином, кальцитонином и комбинацией амилин / кальцитонин, активация рецептора кальцитонина, по-видимому, не имела значения для способности молекулы DACRA к снижению веса и насыщению, которые в основном опосредованы рецептор амилина (155). С другой стороны, кальцитонин и амилин оказывали аддитивное действие на гликемию натощак, предполагая, что активность рецептора кальцитонина может способствовать некоторым улучшениям метаболизма молекул DACRA (155).
Завершено 12-недельное плацебо-контролируемое клиническое исследование фазы II с участием 255 пациентов с диабетом 2 типа, получающих KBP-042 (156), но дальнейшая разработка KBP-042 была прекращена Eli Lilly и Nordic Bioscience, чтобы сосредоточиться на развитие КБП-089, которое, как сообщается, превосходит КБП-042 (157, 158). KBP-089 в настоящее время проходит клинические испытания фазы I на пациентах с диабетом 2 типа (159). На данный момент результаты клинических испытаний молекул DACRA не опубликованы.Таким образом, еще предстоит установить, будут ли многообещающие доклинические результаты, полученные с пептидами DACRA, применяться в возможных стратегиях лечения метаболических заболеваний, таких как ожирение и НАЖБП.
Выводы
В значительной части литературы описаны полезные метаболические эффекты соединений, активирующих рецепторы амилина и кальцитонина по отдельности или в комбинации. Эти эффекты включают потерю массы тела и снижение накопления липидов в печени, которые являются важными краеугольными камнями в лечении ожирения и НАЖБП.Фармацевтические компании реализуют стратегии, основанные на амилине и кальцитонине как на жизнеспособных альтернативах бариатрической хирургии, наиболее эффективному в настоящее время варианту лечения ожирения.
Доклинические и клинические данные подтверждают, что амилин является гормоном против ожирения, тогда как роль кальцитонина при ожирении остается более неопределенной. Тем не менее, кальцитонин лосося, как и новые соединения, такие как аналоги амилина длительного действия и DACRA, демонстрирует потенциал комбинированного агонизма рецепторов амилина и кальцитонина в качестве будущей стратегии лечения ожирения и связанных состояний, таких как НАЖБП.Разделение эффектов этих двойных агонистов через специфические пути амилина и кальцитонина может оказаться трудным без специфических антагонистов рецепторов. Тем не менее, актуально оценивать с клинической точки зрения, чтобы оптимизировать эффекты фармакотерапии, нацеленные на рецепторы амилина и кальцитонина. Будет очень интересно проследить данные клинических программ по исследованию новых соединений амилина и DACRA, поскольку агонисты длительного действия с большей селективностью в отношении рецепторов амилина или кальцитонина могут помочь прояснить это.Однако необходимы специальные исследования для проверки переносимости доклинических данных двойного агонизма амилина и кальцитонина и для определения отдельных и комбинированных физиологических эффектов активности рецепторов амилина и / или кальцитонина у людей.
Вклад авторов
DM подготовил рукопись. AL, TV, FK и JB просмотрели и отредактировали рукопись. Все авторы внесли свой вклад в статью и одобрили представленную версию.
Конфликт интересов
Авторы заявляют, что исследование проводилось в отсутствие каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Ссылки
1. Бузцетти Э., Пинзани М, Цочацис Э.А. Множественный патогенез неалкогольной жировой болезни печени (НАЖБП). Метаболизм (2016) 65: 1038–48. doi: 10.1016 / j.metabol.2015.12.012
PubMed Аннотация | CrossRef Полный текст | Google Scholar
2. Юноси З.М., Кениг А.Б., Абделатиф Д., Фазель Й., Генри Л., Ваймер М. Глобальная эпидемиология неалкогольной жировой болезни печени — метааналитическая оценка распространенности, заболеваемости и исходов. Гепатология (2016) 64: 73–84. doi: 10.1002 / hep.28431
PubMed Аннотация | CrossRef Полный текст | Google Scholar
3. Леони С., Товоли Ф., Неаполь Л., Серио I, Ферри С., Болонди Л. Текущие рекомендации по ведению неалкогольной жировой болезни печени: систематический обзор со сравнительным анализом. World J Gastroenterol (2018) 24: 3361–73. doi: 10.3748 / wjg.v24.i30.3361
PubMed Аннотация | CrossRef Полный текст | Google Scholar
4. Марджот Т., Мула А., Кобболд Дж. Ф., Ходсон Л., Томлинсон Дж. У.Безалкогольная жировая болезнь печени у взрослых: современные концепции этиологии, исходов и лечения. Endocr Ред. (2020) 41: 66–117. doi: 10.1210 / endrev / bnz009
CrossRef Полный текст | Google Scholar
5. Musso G, Cassader M, Rosina F, Gambino R. Влияние текущих методов лечения на заболевания печени, метаболизм глюкозы и сердечно-сосудистый риск при неалкогольной жировой болезни печени (НАЖБП): систематический обзор и метаанализ рандомизированные испытания. Диабетология (2012) 55: 885–904.doi: 10.1007 / s00125-011-2446-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
6. Армстронг MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, et al. Глюкагоноподобный пептид 1 снижает липотоксичность при неалкогольном стеатогепатите. J Hepatol (2016) 64: 399–408. doi: 10.1016 / j.jhep.2015.08.038
PubMed Аннотация | CrossRef Полный текст | Google Scholar
7. Асси Н., Хусейн О., Абасси З. Снижение веса, вызванное орлистатом, устраняет жировую инфильтрацию и улучшает фиброз печени у пациентов с ожирением и неалкогольным стеатогепатитом. Кишечник (2007) 56: 443–4. doi: 10.1136 / gut.2006.106021
PubMed Аннотация | CrossRef Полный текст | Google Scholar
8. Андреассен К.В., Фей М., Хьюлер С.Т., Гайдесен С., Хенриксен Дж. Э., Бек-Нильсен Х. и др. Новый пероральный двойной агонист рецепторов амилина и кальцитонина (KBP-042) оказывает противодействие ожирению и антидиабетический эффект у крыс. Am J Physiol Endocrinol Metab (2014) 307: E24–33. doi: 10.1152 / ajpendo.00121.2014
PubMed Аннотация | CrossRef Полный текст | Google Scholar
9.Gydesen S, Andreassen KV, Hjuler ST, Christensen JM, Karsdal MA, Henriksen K. KBP-088, новый DACRA с пролонгированной активацией рецепторов, превосходит давалинтид с точки зрения эффективности в отношении массы тела. Am J Physiol Endocrinol Metab (2016) 310: E821–7. doi: 10.1152 / ajpendo.00514.2015
PubMed Аннотация | CrossRef Полный текст | Google Scholar
10. Gydesen S, Hjuler ST, Freving Z, Andreassen KV, Sonne N, Hellgren LI, et al. Новый двойной агонист рецепторов амилина и кальцитонина, KBP-089, вызывает потерю веса за счет уменьшения жира, но не мышечной массы, одновременно улучшая пищевые предпочтения. Br J Pharmacol (2017) 174: 591–602. doi: 10.1111 / bph.13723
PubMed Аннотация | CrossRef Полный текст | Google Scholar
11. Hjuler ST, Andreassen KV, Gydesen S, Karsdal MA, Henriksen K. KBP-042 улучшает массу тела и гомеостаз глюкозы с показателями повышенной чувствительности к инсулину независимо от пути введения. Eur J Pharmacol (2015) 762: 229–38. doi: 10.1016 / j.ejphar.2015.05.051
PubMed Аннотация | CrossRef Полный текст | Google Scholar
12.Хьюлер С.Т., Гайдесен С., Андреассен К.В., Педерсен С.Л.К., Хеллгрен Л.И., Карсдал М.А. и др. Двойной агонист рецепторов амилина и кальцитонина KBP-042 повышает чувствительность к инсулину и вызывает потерю веса у крыс с ожирением. Ожирение (2016) 24: 1712–22. doi: 10.1002 / oby.21563
PubMed Аннотация | CrossRef Полный текст | Google Scholar
13. Сонне Н., Ларсен А.Т., Андреассен К.В., Карсдал М.А., Хенриксен К. Двойной агонист рецепторов амилина и кальцитонина, KBP-066, вызывает одинаково сильную потерю веса в широком диапазоне доз, в то время как более высокие дозы могут способствовать дальнейшему улучшению Действие инсулина. J Pharmacol Exp Ther (2020) 373: 92–102. doi: 10.1124 / jpet.119.263723
PubMed Аннотация | CrossRef Полный текст | Google Scholar
14. Европейская ассоциация по изучению печени (EASL), Европейская ассоциация по изучению диабета (EASD), Европейская ассоциация по изучению ожирения (EASO). EASL-EASD-EASO Клинические практические рекомендации по лечению неалкогольной жировой болезни печени. J Hepatol (2016) 64: 1388–402. DOI: 10.1016 / j.jhep.2015.11.004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
15. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. Диагностика и лечение неалкогольной жировой болезни печени: Практическое руководство Американской ассоциации по изучению заболеваний печени. Гепатология (2018) 67: 328–57. doi: 10.1002 / hep.29367
PubMed Аннотация | CrossRef Полный текст | Google Scholar
16. Эслам М., Ньюсом П.Н., Зарин С.К., Ансти К.М., Таргер Г., Ромеро-Гомес М. и др.Новое определение жировой болезни печени, связанной с метаболической дисфункцией: международное экспертное консенсусное заявление. J Hepatol (2020) 73: 202–9. doi: 10.1016 / j.jhep.2020.03.039
PubMed Аннотация | CrossRef Полный текст | Google Scholar
17. Джинджувадиа Р., Антаки Ф., Лохиа П., Лянпунсакул С. Связь между неалкогольной жировой болезнью печени и метаболическими нарушениями у населения США. J Clin Gastroenterol (2017) 51: 160–6. DOI: 10.1097 / MCG.0000000000000666
PubMed Аннотация | CrossRef Полный текст | Google Scholar
18. Дай В., Е Л., Лю А., Вэнь С.В., Дэн Дж., Ву X и др. Распространенность неалкогольной жировой болезни печени у больных сахарным диабетом 2 типа. Med (Балтимор) (2017) 96: e8179. doi: 10.1097 / MD.0000000000008179
CrossRef Полный текст | Google Scholar
19. Котронен А., Пелтонен М., Хаккарайнен А., Севастьянова К., Бергхольм Р., Йоханссон Л. М. и др. Прогнозирование неалкогольной жировой болезни печени и жировой ткани печени с использованием метаболических и генетических факторов. Гастроэнтерология (2009) 137: 865–72. doi: 10.1053 / j.gastro.2009.06.005
PubMed Аннотация | CrossRef Полный текст | Google Scholar
20. Петерсен KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Обращение вспять неалкогольного стеатоза печени, резистентности к инсулину печени и гипергликемии путем умеренного снижения веса у пациентов с диабетом 2 типа. Диабет (2005) 54: 603–8. DOI: 10.2337 / диабет.54.3.603
PubMed Аннотация | CrossRef Полный текст | Google Scholar
21.Suppli MP, Bagger JI, Lund A, Demant M, van Hall G, Strandberg C и др. Устойчивость к глюкагону на уровне обмена аминокислот у лиц с ожирением и стеатозом печени. Диабет (2020) 69: 1090–9. doi: 10.2337 / db19-0715
PubMed Аннотация | CrossRef Полный текст | Google Scholar
23. Мартин BC, Варрам JH, Кролевски AS, Бергман RN, Soeldner JS, Kahn CR. Роль глюкозы и инсулинорезистентности в развитии сахарного диабета 2 типа: результаты последующего 25-летнего исследования. Ланцет (1992) 340: 925–9. doi: 10.1016 / 0140-6736 (92)
-v
PubMed Аннотация | CrossRef Полный текст | Google Scholar
24. Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Инсулинорезистентность при неалкогольной жировой болезни печени. Curr Pharm Des (2010) 16: 1941–51. doi: 10.2174 / 1381612107
875
PubMed Аннотация | CrossRef Полный текст | Google Scholar
25. Нефф К., Ольберс Т., Ле Ру С. Бариатрическая хирургия: проблемы с выбором кандидатов, индивидуальным подходом к лечению и клиническими результатами. BMC Med (2013) 11: 8. doi: 10.1186 / 1741-7015-11-8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
27. Gydesen S, Andreassen KV, Hjuler ST, Hellgren LI, Karsdal MA, Henriksen K. Оптимизация переносимости и эффективности нового двойного агониста рецепторов амилина и кальцитонина KBP-089 посредством увеличения дозы и комбинации с GLP- 1 аналог. Am J Physiol Endocrinol Metab (2017) 313: E598–607. doi: 10.1152 / ajpendo.00419.2016
PubMed Аннотация | CrossRef Полный текст | Google Scholar
28.Харттер Э., Свобода Т., Людвик Б., Шуллер М., Лелл Б., Куэнбург Э. и др. Базальный и стимулированный уровни амилина поджелудочной железы в плазме указывают на его совместную секрецию с инсулином у людей. Диабетология (1991) 34: 52–4. doi: 10.1007 / BF00404025
PubMed Аннотация | CrossRef Полный текст | Google Scholar
29. Батлер П.С., Чоу Дж., Картер В.Б., Ван И-Н, Бу Б-Х, Чанг Д. и др. Влияние приема пищи на концентрацию амилина в плазме у людей с NIDDM и без диабета. Диабет (1990) 39: 752–6.doi: 10.2337 / diab.39.6.752
PubMed Аннотация | CrossRef Полный текст | Google Scholar
31. Boyle CN, Lutz TA, Le Foll C. Amylin — его роль в гомеостатическом и гедоническом контроле за питанием и недавние разработки аналогов амилина для лечения ожирения. Mol Metab (2017) 8: 203–10. doi: 10.1016 / j.molmet.2017.11.009
PubMed Аннотация | CrossRef Полный текст | Google Scholar
32. Янг А.А., Гедулин Б.Р., Ринк Т.Дж. Дозозависимые реакции для замедления опорожнения желудка на модели грызунов глюкагоноподобным пептидом (7-36) Nh3, амилином, холецистокинином и другими возможными регуляторами поглощения питательных веществ. Metab Clin Exp (1996) 45: 1–3. DOI: 10.1016 / s0026-0495 (96)-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
33. Kong M-F, King P, Macdonald IA, Stubbs TA, Perkins AC, Blackshaw PE, et al. Инфузия прамлинтида, аналога человеческого амилина, задерживает опорожнение желудка у мужчин с IDDM. Диабетология (1997) 40: 82–8. doi: 10.1007 / s001250050646
PubMed Аннотация | CrossRef Полный текст | Google Scholar
34. Halawi H, Camilleri M, Acosta A, Vazquez-Roque M, Oduyebo I, Burton D, et al.Связь опорожнения желудка или аккомодации с насыщением, насыщением и постпрандиальными симптомами в состоянии здоровья. Am J Physiol Gastrointest Liver Physiol (2017) 313: G442–7. doi: 10.1152 / ajpgi.00190.2017
PubMed Аннотация | CrossRef Полный текст | Google Scholar
36. Сильвестр Р.А., Родригес-Галлардо Дж., Йодка С., Паркс Д.Г., Питтнер Р.А., Янг А.А. и др. Селективное ингибирование амилином ответа глюкагона на аргинин не относится к поджелудочной железе. Am J Physiol Endocrinol Metab (2001) 280: E443–9.doi: 10.1152 / ajpendo.2001.280.3.E443
PubMed Аннотация | CrossRef Полный текст | Google Scholar
37. McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMP регулируют транспорт и лигандную специфичность рецептора, подобного рецептору кальцитонина. Nature (1998) 393: 333–9. doi: 10.1038 / 30666
PubMed Аннотация | CrossRef Полный текст | Google Scholar
38. Андреассен К.В., Хьюлер С.Т., Фернесс С.Г., Секстон П.М., Кристопулос А., Носин О. и др.Длительная передача сигналов рецепторам кальцитонина лососем, но не кальцитонином человека, выявляет смещение лиганда. PloS One (2014) 9 (3): e
. doi: 10.1371 / journal.pone.00PubMed Аннотация | CrossRef Полный текст | Google Scholar
39. Christopoulos G, Perry KJ, Morfis M, Tilakaratne N, Gao Y, Fraser NJ, et al. Множественные рецепторы амилина возникают в результате взаимодействия белка, изменяющего активность рецептора, с продуктом гена рецептора кальцитонина. Mol Pharmacol (1999) 56: 235–42.doi: 10.1124 / mol.56.1.235
PubMed Аннотация | CrossRef Полный текст | Google Scholar
41. Coester B, Koester-Hegmann C, Lutz TA, Foll CL. Передача сигналов, опосредованная рецептором амилина / кальцитонина, в нейронах POMC влияет на энергетический баланс и двигательную активность у самцов мышей, получавших пищу. Диабет (2020) 69 (6): 1110–25. doi: 10.2337 / db19-0849
PubMed Аннотация | CrossRef Полный текст | Google Scholar
42. Hay DL, Christopoulos G, Christopoulos A, Poyner DR, Sexton PM.Фармакологическая дискриминация рецептора кальцитонина: белковые комплексы, модифицирующие активность рецептора. Mol Pharmacol (2005) 67: 1655–65. doi: 10.1124 / mol.104.008615
PubMed Аннотация | CrossRef Полный текст | Google Scholar
43. Gingell JJ, Burns ER, Hay DL. Активность прамлинтида, амилина крысы и человека, но не Aβ1-42, на рецепторы амилина человека. Эндокринология (2014) 155: 21–6. DOI: 10.1210 / en.2013-1658
PubMed Аннотация | CrossRef Полный текст | Google Scholar
44.Янг А.А., Вайн В., Гедулин Б.Р., Питтнер Р., Джейн С., Гаэта Л.С.Л. и др. Доклиническая фармакология прамлинтида у крыс: сравнение с человеческим и крысиным амилином. Drug Dev Res (1996) 37: 231–48. doi: 10.1002 / (SICI) 1098-2299 (199604) 37: 4 <231 :: AID-DDR5> 3.0.CO; 2-M
CrossRef Полный текст | Google Scholar
46. Арнело Ю., Пермерт Дж., Адриан Т.Э., Ларссон Дж., Вестермарк П., Рейдельбергер Р.Д. Хроническая инфузия островкового амилоидного полипептида вызывает анорексию у крыс. Am J Physiol Regul Integr Comp Physiol (1996) 271: R1654–9.doi: 10.1152 / ajpregu.1996.271.6.R1654
CrossRef Полный текст | Google Scholar
47. Моллет А., Гилг С., Ридигер Т., Лутц Т.А. Инфузия антагониста амилина AC 187 в постремную область увеличивает потребление пищи крысами. Physiol Behav (2004) 81: 149–55. doi: 10.1016 / j.physbeh.2004.01.006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
48. Reidelberger RD, Haver AC, Arnelo U, Smith DD, Schaffert CS, Permert J. Блокада рецепторов амилина стимулирует потребление пищи у крыс. Am J Physiol Regul Integr Comp Physiol (2004) 287: R568–74. doi: 10.1152 / ajpregu.00213.2004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
49. WIELINGA PY, LÖWENSTEIN C, MUFF S, MUNZ M, WOODS SC, LUTZ TA. ЦЕНТРАЛЬНЫЙ АМИЛИН ДЕЙСТВУЕТ В КАЧЕСТВЕ СИГНАЛА ЖИРНОСТИ ДЛЯ КОНТРОЛЯ ВЕСА ТЕЛА И ЭНЕРГЕТИЧЕСКИХ РАСХОДОВ. Physiol Behav (2010) 101: 45–52. doi: 10.1016 / j.physbeh.2010.04.012
PubMed Аннотация | CrossRef Полный текст | Google Scholar
50.Рашинг П.А., Хэган М.М., Сили Р.Дж., Лутц Т.А., Д’Алессио Д.А., Эйр Э.Л. и др. Ингибирование центральной передачи сигналов амилина увеличивает потребление пищи и ожирение у крыс. Эндокринология (2001) 142: 5035. doi: 10.1210 / endo.142.11.8593
PubMed Аннотация | CrossRef Полный текст | Google Scholar
51. Рот Дж. Д., Хьюз Х., Кендалл Э., Барон А. Д., Андерсон К. М.. Эффекты против ожирения бета-клеточного гормона амилина у крыс с ожирением, вызванным диетой: влияние на потребление пищи, массу тела, состав, расход энергии и экспрессию генов. Эндокринология (2006) 147: 5855–64. DOI: 10.1210 / en.2006-0393
PubMed Аннотация | CrossRef Полный текст | Google Scholar
52. Mack C, Wilson J, Athanacio J, Reynolds J, Laugero K, Guss S, et al. Фармакологические действия пептидного гормона амилина в долгосрочном регулировании приема пищи, пищевых предпочтений и массы тела. Am J Physiol Regul Integr Comp Physiol (2007) 293: R1855–63. doi: 10.1152 / ajpregu.00297.2007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
53.Бхавсар С., Уоткинс Дж., Янг А. Синергия между амилином и холецистокинином для ингибирования приема пищи у мышей. Physiol Behav (1998) 64: 557–61. doi: 10.1016 / s0031-9384 (98) 00110-3
PubMed Аннотация | CrossRef Полный текст | Google Scholar
54. Моллет А., Мейер С., Граблер В., Гилг С., Шаррер Е., Лутц Т.А. Эндогенный амилин способствует аноректическому эффекту холецистокинина и бомбезина. Пептиды (2003) 24: 91–8. doi: 10.1016 / s0196-9781 (02) 00280-2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
55.Lutz TA, Del Prete E, Szabady MM, Scharrer E. Ослабление аноректических эффектов глюкагона, холецистокинина и бомбезина антагонистом рецепторов амилина CGRP (8-37). Пептиды (1996) 17: 119–24. doi: 10.1016 / 0196-9781 (95) 02046-2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
56. Лутц Т.А., Чуди С., Рашинг П.А., Шаррер Э. Ослабление аноректических эффектов холецистокинина и бомбезина специфическим антагонистом амилина AC 253. Physiol Behav (2000) 70: 533–6.doi: 10.1016 / s0031-9384 (00) 00302-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
57. West DB, Fey D, Woods SC. Холецистокинин постоянно подавляет размер еды, но не ее потребление у крыс, кормящихся бесплатно. Am J Physiol (1984) 246: R776–87. doi: 10.1152 / ajpregu.1984.246.5.R776
PubMed Аннотация | CrossRef Полный текст | Google Scholar
58. Эйден С., Даниэль С., Стейнбрюк А., Шмидт И., Саймон Э. Кальцитонин лосося — мощный ингибитор приема пищи при нарушении передачи сигналов лептина у лабораторных грызунов. J Physiol (Lond) (2002) 541: 1041–8. doi: 10.1113 / jphysiol.2002.018671
CrossRef Полный текст | Google Scholar
59. Осто М., Вилинга П. Я., Ольха Б., Вальзер Н., Лутц Т. А.. Модуляция насыщающего действия амилина центральным грелином, лептином и инсулином. Physiol Behav (2007) 91: 566–72. doi: 10.1016 / j.physbeh.2007.03.017
PubMed Аннотация | CrossRef Полный текст | Google Scholar
60. Рот Дж. Д., Роланд Б. Л., Коул Р. Л., Треваскис Дж. Л., Вейер С., Кода Дж. Э. и др.Реакция на лептин, восстановленная агонизмом амилина при ожирении, вызванном диетой: данные доклинических и клинических исследований. Proc Natl Acad Sci USA (2008) 105: 7257–62. doi: 10.1073 / pnas.0706473105
PubMed Аннотация | CrossRef Полный текст | Google Scholar
61. Бойл С.Н., Россье М.М., Лутц Т.А. Влияние кормления с высоким содержанием жиров, ожирения, вызванного диетой, и гиперамилинемии на чувствительность к острому амилину. Physiol Behav (2011) 104: 20–8. DOI: 10.1016 / j.physbeh.2011.04.044
PubMed Аннотация | CrossRef Полный текст | Google Scholar
62. Вестермарк П., Вернштедт С., Виландер Э., Слеттен К. Новый пептид в семействе пептидов, родственных гену кальцитонина, как белок амилоидных фибрилл в эндокринной поджелудочной железе. Biochem Biophys Res Commun (1986) 140: 827–31. doi: 10.1016 / 0006-291X (86)
-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
63. Бретертон-Ватт Д., Гилби С.Г., Гатей М.А., Бичем Дж., Макрэ А.Д., Блум С.Р.Для изменения реакции инсулина на внутривенное введение глюкозы у человека необходимы очень высокие концентрации островкового амилоидного полипептида. J Clin Endocrinol Metab (1992) 74: 1032–5. doi: 10.1210 / jcem.74.5.1569151
PubMed Аннотация | CrossRef Полный текст | Google Scholar
64. Бретертон-Ватт Д., Гилби С.Г., Гатей М.А., Бичем Дж., Блум С.Р. Неспособность установить островковый амилоидный полипептид (амилин) в качестве гормона, ингибирующего циркуляцию бета-клеток у человека. Диабетология (1990) 33: 115–7.doi: 10.1007 / BF00401050
PubMed Аннотация | CrossRef Полный текст | Google Scholar
65. Людвик Б., Лелл Б., Харттер Э., Шнак С., Прагер Р. Уменьшение стимулированного высвобождения амилина предшествует нарушению секреции инсулина при диабете II типа. Диабет (1991) 40: 1615–9. doi: 10.2337 / diab.40.12.1615
PubMed Аннотация | CrossRef Полный текст | Google Scholar
66. Эноки С., Мицукава Т., Такемура Дж., Наказато М., Абурая Дж., Тошимори Х. и др. Уровни амилоидного полипептида островков в плазме при ожирении, нарушении толерантности к глюкозе и инсулиннезависимом сахарном диабете. Diabetes Res Clin Pract (1992) 15: 97–102. doi: 10.1016 / 0168-8227 (92)-2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
67. Ханабуса Т., Кубо К., Оки С., Накано И., Окаи К., Санке Т. и др. Секреция островкового амилоидного полипептида (IAPP) островковыми клетками и его концентрация в плазме у пациентов с инсулиннезависимым сахарным диабетом. Diabetes Res Clin Pract (1992) 15: 89–96. doi: 10.1016 / 0168-8227 (92)
-ZPubMed Аннотация | CrossRef Полный текст | Google Scholar
68.Blackard WG, Clore JN, Kellum JM. Секреторные соотношения амилин / инсулин у мужчин с патологическим ожирением: обратная зависимость от скорости исчезновения глюкозы. J Clin Endocrinol Metab (1994) 78: 1257–60. doi: 10.1210 / jcem.78.5.8175987
PubMed Аннотация | CrossRef Полный текст | Google Scholar
69. Каутцки-Виллер А., Томасет К., Пачини Дж., Клоди М., Людвик Б., Стрели С. и др. Роль секреции островкового амилоидного полипептида у инсулинорезистентных людей. Диабетология (1994) 37: 188.doi: 10.1007 / s001250050092
PubMed Аннотация | CrossRef Полный текст | Google Scholar
70. Roth CL, Bongiovanni KD, Gohlke B, Woelfle J. Изменения в динамическом инсулине и секреции желудочно-кишечного гормона у детей с ожирением. J Pediatr Endocrinol Metab (2010) 23: 1299–309. doi: 10.1515 / jpem.2010.204
PubMed Аннотация | CrossRef Полный текст | Google Scholar
71. Ли HJ, Choe YH, Lee JH, Sohn YB, Kim SJ, Park SW, et al. Отсроченный ответ уровней амилина после пероральной глюкозы у детей с синдромом Прадера-Вилли. Yonsei Med J (2011) 52: 257–62. doi: 10.3349 / ymj.2011.52.2.257
PubMed Аннотация | CrossRef Полный текст | Google Scholar
72. Якобсен С.Х., Олесен С.К., Дирксен С., Йоргенсен Н.Б., Бойсен-Мёллер К.Н., Кильгаст Ю. Изменения в ответах на гормоны желудочно-кишечного тракта, чувствительности к инсулину и функции бета-клеток в течение 2 недель после желудочного обходного анастомоза у субъектов, не страдающих диабетом. OBES Surg (2012) 22: 1084–96. doi: 10.1007 / s11695-012-0621-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
73.Beglinger S, Meyer-Gerspach AC, Graf S, Zumsteg U, Drewe J, Beglinger C и др. Влияние пробного завтрака на пищевые реакции гормонов насыщения и их связь с инсулинорезистентностью у тучных подростков. Ожирение (2014) 22: 2047–52. doi: 10.1002 / oby.20805
PubMed Аннотация | CrossRef Полный текст | Google Scholar
74. Клементова М., Тиме Л., Халузик М., Павловикова Р., Хилл М., Пеликанова Т. и др. Растительная пища повышает уровень гормонов желудочно-кишечного тракта и повышает чувство насыщения больше, чем обработанное мясо, соответствующее энергетическим и макроэлементам, у мужчин с диабетом 2 типа, ожирения и здоровых мужчин: рандомизированное перекрестное исследование в трех группах. Питательные вещества (2019) 11 (1): 157. doi: 10.3390 / nu11010157
CrossRef Полный текст | Google Scholar
75. Робертс К.К., Изадпанах А., Ангади С.С., Барнард Р.Дж. Эффекты интенсивной краткосрочной диеты и физических упражнений: сравнение детей с нормальным весом и детей с ожирением. Am J Physiol Regul Integr Comp Physiol (2013) 305: R552–7. doi: 10.1152 / ajpregu.00131.2013
PubMed Аннотация | CrossRef Полный текст | Google Scholar
76. Пибер Т.Р., Ройтельман Дж., Ли Й., Ласки К.Л., Штейн Д.Т.Прямой радиоиммуноанализ плазмы на амилин- (1-37) крыс: концентрации при приобретенном и генетическом ожирении. Am J Physiol Endocrinol Metab (1994) 267: E156–64. doi: 10.1152 / ajpendo.1994.267.1.E156
CrossRef Полный текст | Google Scholar
80. Остин Лос-Анджелес, Heath H, Go VLW. Регулирование секреции кальцитонина у нормального человека путем изменения сывороточного кальция в пределах физиологического диапазона. J Clin Invest (1979) 64: 1721–4. doi: 10.1172 / JCI109636
PubMed Аннотация | CrossRef Полный текст | Google Scholar
81.Копп Д.Х., Кэмерон Е.К., Чейни Б.А., Дэвидсон АГ, Хенце К.Г. Доказательства кальцитонина — нового гормона паращитовидной железы, снижающего содержание кальция в крови. Эндокринология (1962) 70: 638–49. doi: 10.1210 / endo-70-5-638
PubMed Реферат | CrossRef Полный текст | Google Scholar
82. Hurley DL, Tiegs RD, Wahner HW, Heath HI. Минеральная плотность аксиальной и аппендикулярной костей у пациентов с длительным дефицитом или избытком кальцитонина. N Engl J Med (1987) 317: 537–41. DOI: 10.1056 / NEJM198708273170904
PubMed Аннотация | CrossRef Полный текст | Google Scholar
83. Даунс Р.В. мл., Белл Н.Х., Эттингер М.П., Уолш Б.В., Фавус М.Дж., Мако Б. и др. Сравнение алендроната и интраназального кальцитонина для лечения остеопороза у женщин в постменопаузе. J Clin Endocrinol Metab (2000) 85: 1783–8. doi: 10.1210 / jcem.85.5.6606
PubMed Аннотация | CrossRef Полный текст | Google Scholar
84. Lutz TA, Tschudy S, Rushing PA, Scharrer E. Рецепторы амилина опосредуют аноректическое действие кальцитонина лосося (sCT). Пептиды (2000) 21: 233–8. doi: 10.1016 / S0196-9781 (99) 00208-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
85. Сгамбато С., Пассариелло Н., Паолиссо Г., Марано А., Буонинконти Р., Тесауро П. Влияние человеческого кальцитонина (чСТ) на секрецию инсулина, стимулированную глюкозой и аргинином. Acta Diabetes, шир. (1986) 23: 13–22. doi: 10.1007 / BF02581349
CrossRef Полный текст | Google Scholar
86. Джульяно Д., Пассариелло Н., Сгамбато С., Торелла Р., Д’Онофрио Ф.Модуляция кальцитонином секреции инсулина и глюкагона у человека. Am J Physiol Endocrinol Metab (1982) 242: E206–13. doi: 10.1152 / ajpendo.1982.242.3.E206
CrossRef Полный текст | Google Scholar
87. Циглер Р., Беллвинкель С., Шмидтхен Д., Минне Х. Влияние гиперкальциемии, гиперкальциемии и кальцитонина на стимулированную глюкозой секрецию инсулина у человека. Horm Metab Res (1972) 4:60. doi: 10.1055 / s-0028-1097084
PubMed Аннотация | CrossRef Полный текст | Google Scholar
88.Полимерис А, Папапетру П., Папандроулаки Ф., Тану С. Гиперинсулинемия во время перорального теста на толерантность к глюкозе и высокий нормальный уровень кортизола в сыворотке связаны с повышенной секрецией кальцитонина у здоровых субъектов. ГОРМОНЫ (2011) 10: 304–12. doi: 10.14310 / горм.2002.1322
PubMed Аннотация | CrossRef Полный текст | Google Scholar
90. Сираки М., Ито Х., Фудзимаки Х., Хигучи Т. Взаимосвязь между размером тела и минеральной плотностью костей с особым упором на половые гормоны и гормоны, регулирующие кальций, у пожилых женщин. Endocrinol Japonica (1991) 38: 343–9. doi: 10.1507 / endocrj1954.38.343
CrossRef Полный текст | Google Scholar
91. Abbasi A, Corpeleijn E, Postmus D, Gansevoort RT, de Jong PE, Gans ROB и др. Прокальцитонин в плазме крови связан с ожирением, инсулинорезистентностью и метаболическим синдромом. J Clin Endocrinol Metab (2010) 95: E26–31. doi: 10.1210 / jc.2010-0305
PubMed Аннотация | CrossRef Полный текст | Google Scholar
92. Райан Г., Бриско Т.А., Джоб Л.Обзор прамлинтида в качестве дополнительной терапии при лечении диабета 1 и 2 типа. Drug Des Devel Ther (2009) 2: 203–14. doi: 10.2147 / DDDT.S3225
PubMed Аннотация | CrossRef Полный текст | Google Scholar
93. Колберн В.А., Готтлиб А.Б., Кода Дж., Колтерман О.Г. Фармакокинетика и фармакодинамика AC137 (25,28,29 трипро-амилин, человек) после внутривенных болюсных и инфузионных доз у пациентов с инсулинозависимым диабетом. J Clin Pharmacol (1996) 36: 13–24.doi: 10.1002 / j.1552-4604.1996.tb04147.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
94. Smith SR, Blundell JE, Burns C, Ellero C, Schroeder BE, Kesty NC, et al. Лечение прамлинтидом снижает суточное потребление калорий и объем еды, а также улучшает контроль за приемом пищи у субъектов с ожирением: 6-недельное трансляционное исследование. Am J Physiol Endocrinol Metab (2007) 293: E620-7. doi: 10.1152 / ajpendo.00217.2007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
95.Аронн Л., Фуджиока К., Арода В., Чен К., Халсет А., Кести NC и др. Прогрессивное снижение массы тела после лечения аналогом амилина прамлинтидом у субъектов с ожирением: рандомизированное плацебо-контролируемое исследование фазы 2 с увеличением дозы. J Clin Endocrinol Metab (2007) 92: 2977–83. DOI: 10.1210 / jc.2006-2003
PubMed Аннотация | CrossRef Полный текст | Google Scholar
96. Смит С.Р., Аронн Л.Дж., Бернс С.М., Кести NC, Халсет А.Е., Вейер С. Устойчивая потеря веса после 12-месячного лечения прамлинтидом в качестве дополнения к вмешательству в образ жизни при ожирении. Уход за диабетом (2008) 31: 1816–23. doi: 10.2337 / dc08-0029
PubMed Аннотация | CrossRef Полный текст | Google Scholar
97. Foster GD, Wyatt HR, Hill JO, Makris AP, Rosenbaum DL, Brill C, et al. Вес и метаболические показатели после 2 лет диеты с низким содержанием углеводов и диеты с низким содержанием жиров. Ann Internal Med (2010) 153: 147–57. doi: 10.7326 / 0003-4819-153-3-201008030-00005
CrossRef Полный текст | Google Scholar
98. Hollander P, Maggs DG, Ruggles JA, Fineman M, Shen L, Kolterman OG, et al.Влияние прамлинтида на вес у пациентов с избыточным весом и ожирением, леченных инсулином, диабетом 2 типа. Obes Res (2004) 12: 661–8. doi: 10.1038 / oby.2004.76
PubMed Аннотация | CrossRef Полный текст | Google Scholar
99. Холландер П.А., Леви П., Файнман М.С., Маггс Д.Г., Шен Л.З., Штробель С.А. и др. Прамлинтид в качестве дополнения к инсулиновой терапии улучшает долгосрочный гликемический контроль и контроль веса у пациентов с диабетом 2 типа: рандомизированное контролируемое исследование продолжительностью 1 год. Уход за диабетом (2003) 26: 784–90.doi: 10.2337 / diacare.26.3.784
PubMed Аннотация | CrossRef Полный текст | Google Scholar
100. Карл Д., Филис-Цимикас А., Дарсо Т., Лоренци Г., Келлмейер Т., Лутц К. и др. Прамлинтид как дополнение к инсулину у пациентов с диабетом 2 типа в условиях клинической практики: снижение A1C, скачков глюкозы после приема пищи и веса. Diabetes Technol Ther (2007) 9: 191–9. DOI: 10.1089 / dia.2006.0013
PubMed Аннотация | CrossRef Полный текст | Google Scholar
101.Мэггс Д., Шен Л., Штробел С., Браун Д., Колтерман О., Вейер С. Влияние прамлинтида на A1C и массу тела у леченных инсулином афроамериканцев и выходцев из Латинской Америки с диабетом 2 типа: объединенный апостериорный анализ. Метаболизм (2003) 52: 1638–42. doi: 10.1016 / j.metabol.2003.06.003
PubMed Аннотация | CrossRef Полный текст | Google Scholar
102. Ratner RE, Want LL, Fineman MS, Velte MJ, Ruggles JA, Gottlieb A, et al. Дополнительная терапия аналогом амилина прамлинтидом приводит к комбинированному улучшению гликемии и контроля веса у леченных инсулином субъектов с диабетом 2 типа. Diabetes Technol Ther (2002) 4: 51–61. doi: 10.1089 / 1520524
PubMed Аннотация | CrossRef Полный текст | Google Scholar
103. Риддл М., Фриас Дж., Чжан Б., Майер Х., Браун С., Лутц К. и др. Прамлинтид улучшает гликемический контроль и снижает вес у пациентов с диабетом 2 типа, использующих базальный инсулин. Уход за диабетом (2007) 30: 2794–9. doi: 10.2337 / dc07-0589
PubMed Аннотация | CrossRef Полный текст | Google Scholar
104. Томпсон Р.Г., Пирсон Л., Шонфельд С.Л., Колтерман О.Г.Прамлинтид, синтетический аналог амилина человека, улучшает метаболический профиль у пациентов с диабетом 2 типа, получающих инсулин. Прамлинтид в группе диабета 2 типа. Уход за диабетом (1998) 21: 987–93. doi: 10.2337 / diacare.21.6.987
PubMed Аннотация | CrossRef Полный текст | Google Scholar
105. Fineman M, Bahner A, Gottlieb A, Kolterman OG. Влияние шестимесячного приема прамлинтида в качестве дополнения к инсулиновой терапии на метаболический контроль у людей с диабетом 1 типа. Диабет (1999) 48: SA113–3.
Google Scholar
106. Ратнер Р. Э., Дики Р., Файнман М., Мэггс Д. Г., Шен Л., Штробель С. А. и др. Замена амилина прамлинтидом в качестве дополнения к инсулиновой терапии улучшает долгосрочный гликемический контроль и контроль веса при сахарном диабете 1 типа: однолетнее рандомизированное контролируемое исследование. Diabetic Med (2004) 21: 1204–12. doi: 10.1111 / j.1464-5491.2004.01319.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
107.Хаффман Д.М., Маклин Г.В., Сигроув Массачусетс. Непрерывная подкожная инфузионная терапия прамлинтидом у пациентов с диабетом 1 типа: наблюдения пилотного исследования. Endocr Pract (2009) 15: 689–95. doi: 10.4158 / EP09044.ORR1
PubMed Аннотация | CrossRef Полный текст | Google Scholar
108. Кинг А. Минимальное снижение дозы инсулина при терапии прамлинтидом, когда перед лечением установлена почти нормальная гликемия и используется болюс для приема пищи прямоугольной формы. Эндокринная практика (2009) 15: 229–33.doi: 10.4158 / EP.15.3.229
CrossRef Полный текст | Google Scholar
109. Кишияма С.М., Бердик П.Л., Кобри Е.С., Гейдж В.Л., Мессер Л.Х., МакФанн К. Чейз HP. Пилотное испытание домашнего использования прамлинтида у подростков с диабетом 1 типа. Педиатрия (2009) 124: 1344–7. doi: 10.1542 / peds.2008-3750
PubMed Аннотация | CrossRef Полный текст | Google Scholar
110. Ратнер Р., Уайтхаус Ф., Файнман М.С., Штробель С., Шен Л., Мэггс Д.Г. и др. Дополнительная терапия прамлинтидом снижает HbA1c без сопутствующего набора веса и повышенного риска тяжелой гипогликемии у пациентов с диабетом 1 типа, приближающихся к гликемическим целевым показателям. Exp Clin Endocrinol Diabetes (2005) 113: 199–204. doi: 10.1055 / s-2005-837662
PubMed Аннотация | CrossRef Полный текст | Google Scholar
111. Шерр Дж. Л., Патель Н. С., Мишо К. И., Палау-Коллазо М. М., Ван Имя М. А., Тамборлан В. В. и др. Смягчение гликемических экскурсий, связанных с приемом пищи, инсулино-сберегающим способом во время замкнутой доставки инсулина: положительные эффекты дополнительных прамлинтида и лираглутида. Уход за диабетом (2016) 39: 1127–34. doi: 10.2337 / dc16-0089
PubMed Аннотация | CrossRef Полный текст | Google Scholar
112.Whitehouse F, Kruger DF, Fineman M, Shen L, Ruggles JA, Maggs DG, et al. Колтерман О.Г. Рандомизированное исследование и открытое расширение по оценке долгосрочной эффективности прамлинтида в качестве дополнения к инсулиновой терапии при диабете 1 типа. Уход за диабетом (2002) 25: 724–30. doi: 10.2337 / diacare.25.4.724
PubMed Аннотация | CrossRef Полный текст | Google Scholar
113. Уодден Томас А., Уэбб Виктория Л., Моран Кэролайн Х., Бейлер Брук А. Модификация образа жизни при ожирении. Тираж (2012) 125: 1157–70.doi: 10.1161 / CIRCULATIONAHA.111.039453
PubMed Аннотация | CrossRef Полный текст | Google Scholar
114. Брей Г.А., Шателье А., Дункан С., Гринуэй Флорида, Леви Е., Райан Д.Х. и др. Исследовательская группа Программы профилактики диабета. 10-летнее наблюдение за заболеваемостью диабетом и потерей веса в рамках исследования результатов программы профилактики диабета. Ланцет (2009) 374: 1677–86. DOI: 10.1016 / S0140-6736 (09) 61457-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
115.Пи-Суньер Х, Аструп А, Фуджиока К., Гринуэй Ф, Халперн А, Кремпф М. и др. Рандомизированное контролируемое испытание 3,0 мг лираглутида для контроля веса. New Engl J Med (2015) 373: 11–22. doi: 10.1056 / NEJMoa1411892
PubMed Аннотация | CrossRef Полный текст | Google Scholar
116. Салминен П., Хельмио М., Оваска Дж., Джуути А., Лейвонен М., Перомаа-Хаависто П. и др. Эффект лапароскопической рукавной гастрэктомии по сравнению с лапароскопическим обходным желудочным анастомозом по Ру на потерю веса через 5 лет среди пациентов с патологическим ожирением: рандомизированное клиническое испытание SLEEVEPASS. JAMA (2018) 319: 241–54. doi: 10.1001 / jama.2017.20313
PubMed Аннотация | CrossRef Полный текст | Google Scholar
117. Tschöp MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Müller TD, et al. Мономолекулярная полипрагмазия для лечения диабета и ожирения. Cell Metab (2016) 24: 51–62. doi: 10.1016 / j.cmet.2016.06.021
PubMed Аннотация | CrossRef Полный текст | Google Scholar
118. Рот Дж. Д., Коффи Т., Йодка С. М., Майер Х., Атанацио Дж. Р., Мак К. М. и др.Комбинированная терапия амилином и пептидом YY [3–36] у тучных грызунов: анорексигенная синергия и аддитивная потеря веса. Эндокринология (2007) 148: 6054–61. doi: 10.1210 / en.2007-0898
PubMed Аннотация | CrossRef Полный текст | Google Scholar
119. Рот Дж. Д., Д’Суза Л., Гриффин П. С., Атанацио Дж., Треваскис Дж. Л., Назарбаги Р. и др. Взаимодействие амилинэргической и меланокортинергической систем в контроле потребления пищи и массы тела у грызунов. Диабет, ожирение, Metab (2012) 14: 608–15.doi: 10.1111 / j.1463-1326.2012.01570.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
120. Aronne LJ, Halseth AE, Burns CM, Miller S, Shen LZ. Повышенная потеря веса после одновременного приема прамлинтида с сибутрамином или фентермином в многоцентровом исследовании. Ожирение (2010) 18: 1739–46. doi: 10.1038 / oby.2009.478
PubMed Аннотация | CrossRef Полный текст | Google Scholar
121. Ravussin E, Smith SR, Mitchell JA, Shringarpure R, Shan K, Maier H, et al.Повышенная потеря веса с помощью прамлинтида / метрелептина: интегрированный нейрогормональный подход к фармакотерапии ожирения. Obes (Серебряная весна) (2009) 17: 1736–43. doi: 10.1038 / oby.2009.184
CrossRef Полный текст | Google Scholar
122. Bello NT, Kemm MH, Ofeldt EM, Moran TH. Комбинации доз эксендина-4 и кальцитонина лосося вызывают дополнительное и синергетическое снижение потребления пищи у нечеловеческих приматов. Am J Physiol Regul Integr Comp Physiol (2010) 299: R945–52.doi: 10.1152 / ajpregu.00275.2010
PubMed Аннотация | CrossRef Полный текст | Google Scholar
123. Liberini CG, Koch-Laskowski K, Shaulson E, McGrath LE, Lipsky RK, Lhamo R, et al. Комбинированная фармакотерапия амилином / GLP-1 для стимулирования и поддержания длительной потери веса. Sci Rep (2019) 9: 1–11. doi: 10.1038 / s41598-019-44591-8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
124. Novo Nordisk A / S. Исследование безопасности, переносимости, фармакокинетики и фармакодинамики многократного приема NNC0174-0833 в комбинации с семаглутидом у субъектов с избыточным весом или с ожирением (2020).Доступно по адресу: https://clinicaltrials.gov/ct2/show/NCT03600480 (по состоянию на 25 августа 2020 г.).
Google Scholar
125. Мак К.М., Соарес К.Дж., Уилсон Дж. К., Атанацио Дж. Р., Турек В. Ф., Треваскис Дж. Л. и др. Давалинтид (AC2307), новый амилин-миметический пептид: улучшенные фармакологические свойства по сравнению с нативным амилином, что снижает потребление пищи и массу тела. Int J Obes (2010) 34: 385–95. doi: 10.1038 / ijo.2009.238
CrossRef Полный текст | Google Scholar
126. Mack CM, Smith PA, Athanacio JR, Xu K, Wilson JK, Reynolds JM, et al.Глюкорегуляторные эффекты и увеличенная продолжительность действия давалинтида: нового амилиномиметического пептида. Диабет, ожирение, Metab (2011) 13: 1105–13. doi: 10.1111 / j.1463-1326.2011.01465.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
127. Hilton J, Dowton M, Houssami S, Sexton P. Идентификация ключевых компонентов необратимости связывания кальцитонина лосося с рецепторами кальцитонина. J Endocrinol (2000) 166: 213–26. doi: 10.1677 / joe.0.1660213
PubMed Аннотация | CrossRef Полный текст | Google Scholar
128.Закариассен Х.Л., Джон Л.М., Лутц Т.А. Центральный контроль энергетического баланса агонистами рецепторов амилина и кальцитонина и их потенциал для лечения метаболических заболеваний. Basic Clin Pharmacol Toxicol (2020) 127: 163–77. doi: 10.1111 / bcpt.13427
PubMed Аннотация | CrossRef Полный текст | Google Scholar
129. Геррейро Л. Х., Гутеррес MFAN, Мело-Феррейра Б., Эрталь Л. С., Роса М да С., Лоуренсу Д. и др. Получение и характеристика ПЭГилированного амилина. AAPS PharmSciTech (2013) 14: 1083–97.doi: 10.1208 / s12249-013-9987-4
PubMed Аннотация | CrossRef Полный текст | Google Scholar
130. Guterres MFAN, Guerreiro LH, Melo-Ferreira B, Erthal LCS, Lima LMTR. Конъюгирование амилина с метоксилполиэтиленгликолем. Protein Pept Lett (2013) 20: 1264–71. doi: 10.2174 / 0651132099
PubMed Аннотация | CrossRef Полный текст | Google Scholar
131. Kowalczyk R, Brimble MA, Tomabechi Y, Fairbanks AJ, Fletcher M, Hay DL. Конвергентный хемоэнзиматический синтез библиотеки гликозилированных аналогов прамлинтида: взаимосвязь структура-активность для агонизма рецептора амилина. Org Biomol Chem (2014) 12: 8142–51. doi: 10.1039 / c4ob01208a
PubMed Аннотация | CrossRef Полный текст | Google Scholar
132. Nascimento CVMF, Sinezia C, Sisnande T, Lima LMTR, Lacativa PGS. BZ043, новый аналог амилина длительного действия, снижает опорожнение желудка, потребление пищи, гликемию и потребность в инсулине у крыс с индуцированным стрептозотоцином диабетом. Пептиды (2019) 114: 44–9. doi: 10.1016 / j.peptides.2019.04.004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
133.Томабечи Ю., Криппнер Дж., Рендл П.М., Сквайр М.А., Фэрбенкс А.Дж. Гликозилирование прамлинтида: синтетические гликопептиды, которые проявляют активность in vitro и in vivo в качестве агонистов рецептора амилина. Химия (2013) 19: 15084–8. doi: 10.1002 / chem.201303303
PubMed Реферат | CrossRef Полный текст | Google Scholar
134. Yule LR, Bower RL, Kaur H, Kowalczyk R, Hay DL, Brimble MA. Синтез и активность рецептора амилина гликомиметиков прамлинтида с использованием химии кликов. Org Biomol Chem (2016) 14: 5238–45.doi: 10.1039 / c6ob00850j
PubMed Аннотация | CrossRef Полный текст | Google Scholar
136. Брингс А., Боргхардт Дж. М., Скарбалиене Дж., Баадер-Паглер Т., Дерябина М.А., Рист В. и др. Моделирование влияния на потребление энергии и массу тела аналога амилина пролонгированного действия. J Pharmacokinet Pharmacodynamics (2018) 45: 215–33. doi: 10.1007 / s10928-017-9557-6
CrossRef Полный текст | Google Scholar
137. Kalafateli AL, Vestlund J, Raun K, Egecioglu E, Jerlhag E. Влияние селективного агониста амилиновых рецепторов длительного действия на потребление алкоголя, прием пищи и массу тела у самцов и самок крыс. Addict Biol (2020) e12910. doi: 10.1111 / adb.12910
PubMed Аннотация | CrossRef Полный текст | Google Scholar
138. Ново Нордиск, A / S. Исследование фармакокинетических свойств, безопасности и переносимости однократных подкожных доз NNC0174-0833 у мужчин японского и европейского происхождения с нормальным весом, избыточным весом или с ожирением (2019). Доступно по адресу: https://clinicaltrials.gov/ct2/show/NCT03787225 (по состоянию на 25 августа 2020 г.).
Google Scholar
144.Boehringer Ingelheim. Безопасность, переносимость, фармакокинетика и фармакодинамика однократных повышающихся подкожных доз BI 473494 у здоровых мужчин (простой слепой, частично рандомизированный, плацебо-контролируемый параллельный дизайн групп) (2018). Доступно по адресу: https://clinicaltrials.gov/ct2/show/NCT03195088 (по состоянию на 25 августа 2020 г.).
Google Scholar
146. Галанте Л., Хортон Р., Джоплин Г.Ф., Вудхаус, Нью-Джерси, Макинтайр И. Сравнение синтетических кальцитонинов человека, свиньи и лосося у человека и крысы. Clin Sci (1971) 40: 9P.3–10P. doi: 10.1042 / cs040009Pb
CrossRef Полный текст | Google Scholar
147. Йондерко Г., Йондерко К., Голаб Т. Влияние кальцитонина на опорожнение желудка, постпрандиальный гастрин и высвобождение инсулина у пациентов с язвой желудка I типа. Neth J Med (1990) 37: 11–6. doi: 10.1136 / gut.30.4.430
PubMed Аннотация | CrossRef Полный текст | Google Scholar
148. Йондерко К., Конца А., Голаб Т., Йондерко Г. Влияние кальцитонина на объем желчного пузыря у человека. J Gastroenterol Hepatol (1989) 4: 505–11. doi: 10.1111 / j.1440-1746.1989.tb00854.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
151. Feigh M, Henriksen K, Andreassen KV, Hansen C, Henriksen JE, Beck-Nielsen H, et al. Новая пероральная форма кальцитонина лосося улучшает гомеостаз глюкозы и снижает массу тела у крыс с ожирением, вызванным диетой. Диабет, ожирение, Metab (2011) 13: 911–20. doi: 10.1111 / j.1463-1326.2011.01425.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
152.Feigh M, Hjuler ST, Andreassen KV, Gydesen S, Ottosen I., Henriksen JE, et al. Оральный кальцитонин лосося усиливает действие инсулина и метаболизм глюкозы у крыс с ожирением, страдающим стрептозотоцином и диабетом, вызванным диетой. Eur J Pharmacol (2014) 737: 91–6. doi: 10.1016 / j.ejphar.2014.05.016
PubMed Аннотация | CrossRef Полный текст | Google Scholar
154. Tilakaratne N, Christopoulos G, Zumpe ET, Foord SM, Sexton PM. Фенотипы рецептора амилина, полученные из коэкспрессии рецептора кальцитонина человека / RAMP, демонстрируют фармакологические различия, зависящие от изоформы рецептора и окружающей среды клетки-хозяина. J Pharmacol Exp Ther (2000) 294: 61–72.
PubMed Аннотация | Google Scholar
155. Ларсен А.Т., Сонне Н., Андреассен К.В., Карсдал М.А., Хенриксен К. Рецептор кальцитонина играет главную роль в регуляции глюкозы как функция терапии двойным амилином и агонистом рецептора кальцитонина. J Pharmacol Exp Ther (2020) 374: 74–83. doi: 10.1124 / jpet.119.263392
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Логарифмическая оптимальность методом наименьших квадратов среднего геометрического весовых векторов, вычисленных по всем остовным деревьям для (неполных) полных матриц парных сравнений
[3] Choo, E.У., Уэдли, У. (2004): Общая структура для получения значений предпочтений из матриц парных сравнений
, Computers & Operations Research, 31 (6) 893–908
[4] Чу, ATW, Kalaba, RE, Spingarn, K. (1979) : Сравнение двух методов определения
определения весов принадлежности к нечетким множествам, Journal of Optimization Theory and Appli-
катионов, 27 (4) 531–538
[5] Crawford, G., Williams, К. (1985): Заметка об анализе матриц субъективных суждений,
Journal of Mathematical Psychology 29 (4) 387–405
[6] Fichtner, J.(1984). Некоторые мысли о математике аналитической иерархии Pro-
cess. Отчет 8403, Universit¨at der Bundeswehr M¨unchen, Fakult¨at f¨ur Informatik, Institut
f¨ur Angewandte Systemforschung und Operations Research, Werner-Heisenberg-Weg 39,
D-8014 Neubiberg, F.R.G. 1984.
[7] Fichtner, J. (1986). О получении векторов приоритета из матриц парных сравнений.
Науки о социально-экономическом планировании, 20 (6) 341–345
[8] Голани, Б., Кресс, М. (1993): Многокритериальная оценка методов получения весов
из матриц шкалы отношений, European Journal of Operational Research, 69 (2) 210–220
[9] Harker, P.T. (1987): Неполные парные сравнения в процессе аналитической иерархии,
Математическое моделирование 9 (11) 837–848
[10] де Йонг П. (1984): Статистический подход к методам масштабирования Саати для приоритетов, журнал
математической психологии 28 (4) 467–478
[11] Квесилевич М.(1996): Логарифмический метод наименьших квадратов и обобщенная псевдообратная величина
при оценке отношений, Европейский журнал операционных исследований 93 (3) 611–619
[12] Ланди, М., Сирадж, С., Греко, С. ( 2017): Математическая эквивалентность «охватывающего дерева
» и векторов предпочтений среднего геометрического ряда и его значение для анализа предпочтений,
European Journal of Operational Research 257 (1) 197–208
[13] Saaty, TL (1977): метод масштабирования приоритетов в иерархических структурах, журнал
Mathematical Psychology 15 (3) 234–281
[14] Siraj, S., Михайлов, Л., Кин, Дж. (2012): Перечисление всех покрывающих деревьев для попарных сравнений
, Computers & Operations Research, 39 (2) 191–199
[15] Сирадж, С., Михайлов, Л., Кин, Дж. А. (2012): Исправление к «Перечислению всех покрывающих
деревьев для попарных сравнений [Comput. Опер. Res. 39 (2012) 191-199] », Computers & Op-
erations Research, 39 (9) page 2265
[16] Цыганок В. (2000): Комбинаторный метод попарных сравнений с обратной связью, Data
Recording , Хранение и обработка 2: 92–102 (на украинском языке).
[17] Цыганок В. (2010): Исследование эффективности агрегирования экспертных оценок
, полученных методом попарного сравнения, Математическое и компьютерное моделирование, 52 (3-
4) 538–54
8
Миозит с тельцами включения: обновленная информация о патогенезе и лечении
Callan A, Capkun G, Vasanthaprasad V, Freitas R, Needham M (2017) Систематический обзор и метаанализ исследований распространенности спорадического миозита с тельцами включения.J Neuromuscul Dis 4: 127–137
Статья PubMed Google Scholar
Capkun G, Callan A, Tian H, Wei Z, Zhao C, Agashivala N, Barghout V (2017) Бремя болезни и использование ресурсов здравоохранения у пациентов в США со спорадическим миозитом с тельцами включения. Muscle Nerve 56: 861–867
Статья PubMed Google Scholar
Димачкие М.М., Барон Р.Дж. (2014) Миозит с включенными тельцами.Neurol Clin 32: 629–646, vii
Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ (1989) Миозит с включенными тельцами. Наблюдения у 40 больных. Brain 112 (Pt 3): 727–747
Статья PubMed Google Scholar
Oh TH, Brumfield KA, Hoskin TL, Stolp KA, Murray JA, Basford JR (2007) Дисфагия при воспалительной миопатии: клинические характеристики, стратегии лечения и исход у 62 пациентов. Mayo Clin Proc 82: 441–447
Статья PubMed Google Scholar
Ghosh PS, Milone M (2015) Camptocormia как проявление спектра миопатических расстройств. Muscle Nerve 52: 1008–1012
Статья PubMed Google Scholar
Ghosh PS, Laughlin RS, Engel AG (2014) Миозит с включенными тельцами, проявляющийся диплегией лица. Muscle Nerve 49: 287–289
Статья. CAS PubMed Google Scholar
Voermans NC, Vaneker M, Hengstman GJD, ter Laak HJ, Zimmerman C, Schelhaas HJ, Zwarts MJ (2004) Первичная дыхательная недостаточность при миозите с тельцами включения. Неврология 63: 2191–2192
Статья CAS PubMed Google Scholar
Чахин Н., Энгель А.Г. (2008) Корреляция мышечной биопсии, клинического течения и исходов при ПМ и спорадических IBM. Неврология 70: 418–424
Статья. PubMed Google Scholar
Brady S, Squier W, Sewry C, Hanna M, Hilton-Jones D, Holton JL (2014) Ретроспективное когортное исследование, определяющее основные патологические особенности, полезные для диагностики миозита с тельцами включения. BMJ Open 4: e004552
Статья PubMed PubMed Central Google Scholar
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR (2002) Диагностические критерии нарушений дыхательной цепи у взрослых и детей.Неврология 59: 1406–1411
Статья. CAS PubMed Google Scholar
Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, Rowland LP (1995) Миозит с включенными тельцами и миопатии. Ann Neurol 38: 705–713
Статья CAS PubMed Google Scholar
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG (2010) Миозит с включенными тельцами: MRC Center for Neuromuscular Diseases, семинар IBM, Лондон, 13 июня 2008 г.Neuromuscul Disord 20: 142–147
Статья CAS PubMed Google Scholar
Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA (2009) Саркоплазматическое перераспределение ядерного TDP-43 при миозите с тельцами включения. Muscle Nerve 40: 19–31
Статья CAS PubMed PubMed Central Google Scholar
Nogalska A, Terracciano C, D’Agostino C, King Engel W., Askanas V (2009) p62 / SQSTM1 сверхэкспрессируется и заметно накапливается во включениях спорадических мышечных волокон миозита с тельцами включения и может помочь дифференцировать его. от полимиозита и дерматомиозита.Acta Neuropathol 118: 407–413
Статья CAS PubMed Google Scholar
Dubourg O, Wanschitz J, Maisonobe T., Béhin A, Allenbach Y, Herson S, Benveniste O (2011) Диагностическая ценность маркеров мышечной дегенерации при спорадическом миозите с тельцами включения. Acta Myol 30 (2): 103–8
Rose MR (2013) 188-й международный семинар ENMC: миозит с тельцами включения, 2–4 декабря 2011 г., Наарден, Нидерланды.Нейромышечные расстройства 23: 1044–1055
Статья CAS PubMed Google Scholar
Salajegheh M, Lam T, Greenberg SA (2011) Аутоантитела против мышечного белка 43 кДа при миозите с тельцами включения. PLoS One 6: e20266
Артикул CAS PubMed PubMed Central Google Scholar
Pluk H, van Hoeve BJA, van Dooren SHJ, et al (2013) Аутоантитела к цитозольной 5′-нуклеотидазе 1A при миозите с тельцами включения.Ann Neurol 73: 397–407
Статья CAS PubMed Google Scholar
Ларман Б.Х., Саладжеге М., Назарено Р. и др. (2013) Аутоиммунитет цитозольной 5′-нуклеотидазы 1A при спорадическом миозите с тельцами включения. Ann Neurol 73: 408–418
Статья CAS PubMed Google Scholar
Tawara N, Yamashita S, Zhang X, et al (2017) Патомеханизмы аутоантител против цитозольной 5′-нуклеотидазы 1A при спорадическом миозите с тельцами включения.Ann Neurol 81: 512–525
Статья CAS PubMed Google Scholar
Lloyd TE, Christopher-Stine L, Pinal-Fernandez I, Tiniakou E, Petri M, Baer A, Danoff SK, Pak K, Casciola-Rosen LA, Mammen AL (2016) Цитозольная 5′-нуклеотидаза 1A как мишень циркулирующих аутоантител при аутоиммунных заболеваниях Arthritis Care Res (Hoboken) 68: 66–71
Статья CAS Google Scholar
Goyal NA, Cash TM, Alam U, Enam S, Tierney P, Araujo N, Mozaffar FH, Pestronk A, Mozaffar T (2016) Серопозитивность антител NT5c1A при спорадическом миозите с тельцами включения предсказывает более тяжелое моторное, бульбарное и респираторное поражение. J Neurol Neurosurg Psychiatry 87: 373–378
Статья CAS PubMed Google Scholar
Lilleker JB, Rietveld A, Pye SR, et al (2017) Профиль аутоантител к цитозольным 5′-нуклеотидазе 1A и клинические характеристики при миозите с телеками включения.Ann Rheum Dis 76: 862–868
Статья CAS PubMed PubMed Central Google Scholar
Herbert MK, Stammen-Vogelzangs J, Verbeek MM, et al (2016) Болезненная специфичность аутоантител к цитозольной 5′-нуклеотидазе 1A при спорадическом миозите с тельцами включения по сравнению с известными аутоиммунными заболеваниями. Ann Rheum Dis 75: 696–701
Статья CAS PubMed Google Scholar
Kazamel M, Sorenson EJ, Milone M (2016) Клинические и электрофизиологические данные при наследственной миопатии с тельцами включения по сравнению со спорадическим миозитом с тельцами включения. J Clin Neuromuscul Dis 17: 190–196
Статья PubMed Google Scholar
Бадрисинг У.А., Маат-Шиман М., Ван Дуинен С.Г. и др. (2000) Эпидемиология миозита с тельцами включения в Нидерландах: общенациональное исследование. Неврология 55: 1385–1387
Статья. CAS PubMed Google Scholar
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG (2010) Миозит с включенными тельцами. Neuromuscul Disord 20: 142–147
Статья CAS PubMed Google Scholar
Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA (2014) Оценка и построение диагностических критериев для миозита с включенными телами. Неврология 83: 426–433
Статья. PubMed PubMed Central Google Scholar
Jones KL, Sejersen T, Amato AA, Hilton-Jones D, Schmidt J, Wallace AC, Badrising UA, Rose MR, IBM Guideline Development Group (2016) Протокол для разработки клинических руководств по миозиту с телец включения. Muscle Nerve 53: 503–507
Статья PubMed PubMed Central Google Scholar
Энгель А.Г., Арахата К. (1984) Анализ моноклональных антител мононуклеарных клеток при миопатиях. II: фенотипы аутоинвазивных клеток при полимиозите и миозите с тельцами включения.Ann Neurol 16: 209–215
Статья CAS PubMed Google Scholar
Амемия К., Грейнджер Р.П., Далакас М.К. (2000) Клональное ограничение экспрессии Т-клеточного рецептора инфильтрацией лимфоцитов при миозите с тельцами включения сохраняется с течением времени. Исследования при повторных биопсиях мышц. Brain 123 (Pt 10): 2030–2039
Статья PubMed Google Scholar
Salajegheh M, Rakocevic G, Raju R, Shatunov A, Goldfarb LG, Dalakas MC (2007) Профилирование рецепторов Т-клеток в мышцах и лимфоцитах крови при спорадическом миозите с тельцами включения.Неврология 69: 1672–1679
Статья. CAS PubMed Google Scholar
Tateyama M, Fujihara K, Misu T., Itoyama Y (2009) CCR7 + миелоидные дендритные клетки вместе с CCR7 + T-клетками и CCR7 + макрофагами вторгаются в CCL19 + ненекротические мышечные волокна при миозите с телец включения. J Neurol Sci 279: 47–52
Статья CAS PubMed Google Scholar
Гринберг С.А., Пинкус Г.С., Амато А.А., Пинкус Дж.Л. (2007) Миелоидные дендритные клетки при миозите и полимиозите с тельцами включения.Muscle Nerve 35: 17–23
Статья. CAS PubMed Google Scholar
Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, Bregoli LS, O’Connor KC, Amato AA, Amato AA (2005) Плазматические клетки в мышцах при миозите и полимиозите с тельцами включения . Неврология 65: 1782–1787
Статья. CAS PubMed Google Scholar
Bradshaw EM, Orihuela A, McArdel SL, Salajegheh M, Amato AA, Hafler DA, Greenberg SA, O’Connor KC (2007) При воспалительных миопатиях присутствует местный антиген-управляемый гуморальный ответ.J Immunol 178: 547–556
Статья CAS PubMed Google Scholar
Uruha A, Noguchi S, Hayashi YK, Tsuburaya RS, Yonekawa T., Nonaka I, Nishino I (2016) Инфекция вируса гепатита C при миозите с тельцами включения. Неврология 86: 211–217
Статья. CAS PubMed Google Scholar
Alverne ARSM, Marie SKN, Levy-Neto M, de Souza FHC, de Carvalho MS, Shinjo SK (2013) Миозит с включенными тельцами: серия из 30 случаев из бразильского специализированного центра.Acta Reumatol Port 38: 179–185
PubMed Google Scholar
Куплер Э.Дж., Леон-Монзон М., Миллер Дж., Семино-Мора С., Андерсон Т.Л., Далакас М.С. (1996) Миозит с включенными тельцами у пациентов, инфицированных ВИЧ-1 и HTLV-1. Brain 119 (Pt 6): 1887–1893
Статья PubMed Google Scholar
Dalakas MC, Rakocevic G, Shatunov A, Goldfarb L, Raju R, Salajegheh M (2007) Миозит с тельцами включения с инфекцией вируса иммунодефицита человека: четыре случая с клональной экспансией вирусоспецифичных Т-клеток.Ann Neurol 61: 466–475
Статья CAS PubMed Google Scholar
Гринберг С.А., Пинкус Дж. Л., Амато А.А., Кристенсен Т., Дорфман Д.М. (2016) Ассоциация миозита с тельцами включения с Т-клеточным большим гранулярным лимфоцитарным лейкозом. Мозг 139: 1348–1360
Статья PubMed Google Scholar
Bareau B, Rey J, Hamidou M, et al (2010) Анализ французской когорты пациентов с лейкемией крупных гранулярных лимфоцитов: отчет о 229 случаях.Haematologica 95: 1534–41
Статья CAS PubMed PubMed Central Google Scholar
Hohlfeld R, Schulze-Koops H (2016) Цитотоксические Т-клетки не работают при миозите с тельцами включения. Мозг 139: 1312–1314
Статья PubMed Google Scholar
Lamy T, Moignet A, Loughran TP (2017) Лейкоз LGL: от патогенеза к лечению. Кровь 129: 1082–1094
Статья CAS PubMed Google Scholar
Зануссо Г., Ваттеми Г., Феррари С. и др. (2001) Повышенная экспрессия нормальной клеточной изоформы прионного белка при миозите с тельцами включения, воспалительных миопатиях и атрофии денервации. Brain Pathol 11: 182–189
Статья CAS PubMed Google Scholar
Асканас В., Энгель В.К., Альварес Р.Б., Гленнер Г.Г. (1992) иммунореактивность бета-амилоидного белка в мышцах пациентов с миозитом с тельцами включения. Lancet (Лондон, Англия) 339: 560–561
Статья CAS Google Scholar
Mendell JR, Sahenk Z, Gales T, Paul L (1991) Амилоидные волокна при миозите с тельцами включения. Новые открытия позволяют лучше понять природу волокон. Arch Neurol 48: 1229–1234
Артикул CAS PubMed Google Scholar
Каталон-Гарсия М., Гаррабу Дж., Морен С. и др. (2015) Уровни BACE-1, PS-1 и sAPPβ повышены в плазме у пациентов со спорадическим миозитом с тельцами включения: суррогатные биомаркеры воспалительных миопатий.Mol Med 21: 1
Статья CAS Google Scholar
Nogalska A, D’Agostino C, Engel WK, Klein WL, Askanas V (2010) Новая демонстрация олигомеров амилоида-β в мышечных волокнах спорадического миозита с тельцами включения. Acta Neuropathol 120: 661–666
Статья CAS PubMed Google Scholar
Саркози Э., Асканас В., Джонсон С.А., Энгель В.К., Альварес Р.Б. (1993) мРНК белка-предшественника бета-амилоида увеличивается в мышцах, пораженных миозитом с тельцами включения.Нейроотчет 4: 815–818
Статья CAS PubMed Google Scholar
Abdo WF, van Mierlo T, Hengstman GJ, Schelhaas HJ, van Engelen BG, Verbeek MM (2009) Повышенное содержание белка амилоида-бета42 в плазме при спорадическом миозите с тельцами включения. Acta Neuropathol 118: 429–431
Статья PubMed PubMed Central Google Scholar
Nogalska A, D’Agostino C, Engel WK, Cacciottolo M, Asada S, Mori K, Askanas V (2015) Активация развернутого белкового ответа при спорадическом миозите с тельцами включения, но не при наследственном GNE миопатия с тельцами включения.J Neuropathol Exp Neurol 74: 538–546
Статья CAS PubMed PubMed Central Google Scholar
Finley D (2009) Распознавание и процессинг конъюгатов убиквитин-белок протеасомой. Annu Rev Biochem 78: 477–513
Статья CAS PubMed PubMed Central Google Scholar
Fratta P, Engel WK, McFerrin J, Davies KJA, Lin SW, Askanas V (2005) Ингибирование протеасом и образование агресом при спорадическом миозите с телец включения и в культивируемых мышечных волокнах человека с избыточной экспрессией белка-предшественника амилоида .Am J Pathol 167: 517–526
Статья CAS PubMed PubMed Central Google Scholar
Фукухара Н., Кумамото Т., Цубаки Т. (1980) Вакуоли с краями. Acta Neuropathol 51: 229–235
Статья CAS PubMed Google Scholar
Güttsches A-K, Brady S, Krause K, et al (2017) Протеомика окаймленных вакуолей определяет новый аллель риска при миозите с тельцами включения.Ann Neurol 81: 227–239
Статья CAS PubMed PubMed Central Google Scholar
Ju J-S, Varadhachary AS, Miller SE, Weihl CC (2010) Количественное определение «аутофагического потока» в зрелых скелетных мышцах Autophagy 6: 929–935
Article CAS PubMed PubMed Central Google Scholar
Ching JK, Elizabeth S. V., Ju J-S, Lusk C, Pittman SK, Weihl CC (2013) дисфункция mTOR способствует патологии вакуолей и слабости при миопатии с тельцами включения, связанной с валозин-содержащими белками.Hum Mol Genet 22: 1167–1179
Статья CAS PubMed Google Scholar
Nalbandian A, Llewellyn KJ, Nguyen C, Yazdi PG, Kimonis VE (2015) Рапамицин и хлорохин: эффекты in vitro и in vivo модифицирующих аутофагию препаратов показывают многообещающие результаты в мультисистемной протеинопатии, содержащей валозин. PLoS One 10: e0122888
Статья CAS PubMed PubMed Central Google Scholar
Nogalska A, D’Agostino C, Terracciano C, Engel WK, Askanas V (2010) Нарушение аутофагии при спорадическом миозите с телами включения и в культивируемых мышечных волокнах человека, вызванных стрессом эндоплазматического ретикулума. Am J Pathol 177: 1377–1387
Статья CAS PubMed PubMed Central Google Scholar
Lünemann JD, Schmidt J, Dalakas MC, Münz C (2007) Макроаутофагия как патомеханизм спорадического миозита с тельцами включения.Аутофагия 3: 384–386
Статья. PubMed Google Scholar
Кумамото Т., Уэяма Х, Цумура Х, Тоошима И., Цуда Т. (2004) Экспрессия белков и генов, связанных с лизосомами, в скелетных мышцах миозита с тельцами включения. Acta Neuropathol 107: 59–65
Статья CAS PubMed Google Scholar
Seibenhener ML, Babu JR, Geetha T., Wong HC, Krishna NR, Wooten MW (2004) Секестосома 1 / p62 представляет собой белок, связывающий цепь полиубиквитина, участвующий в деградации протеасомы убиквитина.Mol Cell Biol 24: 8055–8068
Статья CAS PubMed PubMed Central Google Scholar
Nakano S, Oki M, Kusaka H (2017) Роль p62 / SQSTM1 в спорадическом миозите с тельцами включения. Нейромышечное расстройство 27: 363–369
Статья PubMed Google Scholar
Kaufman RJ (1999) Передача сигналов стресса из просвета эндоплазматического ретикулума: координация контроля транскрипции и трансляции генов.Genes Dev 13: 1211–1233
Статья CAS PubMed Google Scholar
Brown IR (2007) Белки теплового шока и защита нервной системы. Ann N Y Acad Sci 1113: 147–158
Статья CAS PubMed Google Scholar
Cacciottolo M, Nogalska A, D’Agostino C, Engel WK, Askanas V (2013) Компоненты аутофагии, опосредованные шаперонами, активируются в спорадических мышечных волокнах миозита с тельцами включения.Neuropathol Appl Neurobiol 39: 750–761
Статья CAS PubMed Google Scholar
Nogalska A, Engel WK, McFerrin J, Kokame K, Komano H, Askanas V (2006) Гомоцистеин-индуцированный белок эндоплазматического ретикулума (Herp) активируется при спорадическом миозите с тельцами включения и при стрессе эндоплазматического ретикулума -индуцированные культивируемые мышечные волокна человека. J Neurochem 96: 1491–1499
Статья CAS PubMed Google Scholar
Banwell BL, Engel AG (2000) Иммунолокализация AlphaB-кристаллина позволяет по-новому взглянуть на миозит с тельцами включения. Неврология 54: 1033–1041
Статья. CAS PubMed Google Scholar
Wójcik S, Engel WK, McFerrin J, Askanas V (2005) Миостатин повышен и образует комплексы с амилоидом-β в спорадических мышечных волокнах миозита с тельцами включения. Acta Neuropathol 110: 173–177
Статья CAS PubMed Google Scholar
Nogalska A, Wojcik S, King Engel W, McFerrin J, Askanas V (2007) Стресс эндоплазматического ретикулума индуцирует белок-предшественник миостатина и NF-κB в культивируемых мышечных волокнах человека: отношение к миозиту с телец включения. Exp Neurol 204: 610–618
Артикул CAS PubMed Google Scholar
Sachdev R, Kappes-Horn K, Paulsen L, Duernberger Y, Pleschka C, Denner P, Kundu B, Reimann J, Vorberg I (2018) Стресс эндоплазматического ретикулума индуцирует высокомолекулярные агрегаты миостатина и нарушает зрелый миостатин секреция.Mol Neurobiol. DOI: https://doi.org/10.1007/s12035-018-0997-9
Gonzalez-Cadavid NF, Bhasin S (2004) Роль миостатина в метаболизме. Curr Opin Clin Nutr Metab Care 7: 451–457
Артикул CAS PubMed Google Scholar
Каталан-Гарсия М., Гаррабу Дж., Морен С. и др. (2016) Нарушения митохондриальной ДНК и нарушение регуляции экспрессии окислительного фосфорилирования и белков слияния митохондрий при спорадическом миозите с тельцами включения.Clin Sci 130: 1741–1751
Статья CAS PubMed Google Scholar
Rygiel KA, Tuppen HA, Grady JP, Vincent A, Blakely EL, Reeve AK, Taylor RW, Picard M, Miller J, Turnbull DM (2016) Комплексные перестройки митохондриальной ДНК в отдельных клетках от пациентов со спорадическим включением миозит тела. Nucleic Acids Res 44: 5313–5329
Статья CAS PubMed PubMed Central Google Scholar
Bhattarai S, Ghannam K, Krause S, et al (2016) Иммунопротеасомы являются ключом к регулированию миокинов и экспрессии MHC класса I при идиопатических воспалительных миопатиях. J Autoimmun 75: 118–129
Статья CAS PubMed Google Scholar
Fréret M, Drouot L, Obry A, Ahmed-Lacheheb S, Dauly C, Adriouch S, Cosette P, Authier FJ, Boyer O (2013) Сверхэкспрессия MHC класса I в мышцах мышей с дефицитом лимфоцитов вызывает тяжелая миопатия с индукцией развернутого белкового ответа.Am J Pathol 183: 893–904
Статья CAS PubMed Google Scholar
Ахмед М., Мачадо П.М., Миллер А. и др. (2016) Нацеленность на гомеостаз белков при спорадическом миозите с тельцами включения. Sci Transl Med 8: 331ra41–331ra41
Статья CAS PubMed PubMed Central Google Scholar
Schmidt J, Barthel K, Wrede A, Salajegheh M, Bähr M, Dalakas MC (2008) Взаимосвязь воспаления и APP в sIBM: IL-1 beta вызывает накопление бета-амилоида в скелетных мышцах.Мозг 131: 1228–1240
Статья PubMed PubMed Central Google Scholar
Adams V, Nehrhoff B, Späte U, Linke A, Schulze PC, Baur A, Gielen S, Hambrecht R, Schuler G (2002) Индукция экспрессии iNOS в скелетных мышцах посредством активации IL-1beta и NFkappaB: исследование in vitro и in vivo. Cardiovasc Res 54: 95–104
Статья CAS PubMed Google Scholar
Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW, Turnbull DM (2015) Митохондриальные и воспалительные изменения при спорадическом миозите с тельцами включения. Neuropathol Appl Neurobiol 41: 288–303
Статья CAS PubMed PubMed Central Google Scholar
Rojana-udomsart A, Bundell C, James I, Castley A, Martinez P, Christiansen F, Hollingsworth P, Mastaglia F (2012) Частота аутоантител и корреляция с генотипом HLA-DRB1 при спорадическом миозите с тельцами включения ( s-IBM): исследование контроля населения.J Neuroimmunol 249: 66–70
Статья CAS PubMed Google Scholar
Rothwell S, Cooper RG, Lundberg IE, et al (2017) Иммунный анализ при спорадическом миозите с тельцами включения выявляет гетерогенность HLA-DRB1 аминокислот по всему спектру миозита. Arthritis Rheumatol 69: 1090–1099
Статья CAS PubMed PubMed Central Google Scholar
Mastaglia FL, Needham M, Scott A, et al (2009) Спорадический миозит с тельцами включения: взаимодействия аллелей HLA-DRB1 влияют на риск заболевания и клинический фенотип. Neuromuscul Disord 19: 763–765
Статья PubMed Google Scholar
Rojana-udomsart A, James I, Castley A, et al (2012) Генотипирование HLA-DRB1 с высоким разрешением в когорте австралийских телец включения (s-IBM): анализ аллелей, связанных с заболеванием, и диплотипы.J Neuroimmunol 250: 77–82
Статья CAS PubMed Google Scholar
Weihl CC, Baloh RH, Lee Y, Chou TF, Pittman SK, Lopate G, Allred P, Jockel-Balsarotti J, Pestronk A, Harms MB (2015) Целевое секвенирование и идентификация генетических вариантов при спорадическом включении миозит тела. Neuromuscul Disord 25: 289–296
Статья PubMed PubMed Central Google Scholar
Gang Q, Bettencourt C, Machado PM и др. (2016) Редкие варианты генов SQSTM1 и VCP и риск спорадического миозита с тельцами включения. Neurobiol Aging 47: 218.e1–218.e9
Статья CAS Google Scholar
Gang Q, Bettencourt C, Machado PM, et al (2015) Эффекты интронного полиморфизма в генотипах TOMM40 и APOE при спорадическом миозите с тельцами включения. Neurobiol Aging 36: 1766.e1–1766.e3
Статья CAS Google Scholar
Mastaglia FL, Rojana-udomsart A, James I, et al (2013) Полиморфизм в гене TOMM40 изменяет риск развития спорадического миозита с тельцами включения и возраст появления симптомов. Нейромышечные расстройства 23: 969–974
Статья CAS PubMed Google Scholar
Hansson Petersen CA, Alikhani N, Behbahani H, et al (2008) Бета-амилоидный пептид импортируется в митохондрии через механизм импорта TOM и локализуется в митохондриальных кристах.Proc Natl Acad Sci U S A 105: 13145–13150
Статья PubMed PubMed Central Google Scholar
Wild P, McEwan DG, Dikic I (2014) Краткий обзор взаимодействия LC3. J Cell Sci 127: 3–9
Статья CAS PubMed Google Scholar
Leff RL, Miller FW, Hicks J, Fraser DD, Plotz PH (1993) Лечение миозита с тельцами включения: ретроспективный обзор и рандомизированное проспективное исследование иммуносупрессивной терапии.Медицина (Балтимор) 72: 225–235
Статья CAS Google Scholar
Amato AA, Barohn RJ, Jackson CE, Pappert EJ, Sahenk Z, Kissel JT (1994) Миозит с включенными тельцами: лечение внутривенным иммуноглобулином. Неврология 44: 1516–1518
Статья. CAS PubMed Google Scholar
Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K (1997) Лечение миозита с тельцами включения с помощью IVIg: двойное слепое плацебо-контролируемое исследование.Неврология 48: 712–716
Статья. CAS PubMed Google Scholar
Walter MC, Lochmüller H, Toepfer M, Schlotter B, Reilich P, Schröder M, Müller-Felber W., Pongratz D (2000) Высокодозная иммуноглобулиновая терапия при спорадическом миозите с тельцами включения: двойной слепой, плацебо-контролируемое исследование. J Neurol 247: 22–28
Статья CAS PubMed Google Scholar
Badrising UA, Maat-Schieman MLC, Ferrari MD, et al (2002) Сравнение прогрессирования слабости при миозите с тельцами включения во время лечения метотрексатом или плацебо. Ann Neurol 51: 369–372
Статья CAS PubMed Google Scholar
Barohn RJ, Amato AA, Sahenk Z, Kissel JT, Mendell JR (1995) Миозит с включенными тельцами: объяснение плохой реакции на иммуносупрессивную терапию. Неврология 45: 1302–1304
Статья. CAS PubMed Google Scholar
Cherin P, Pelletier S, Teixeira A, Laforet P, Simon A, Herson S, Eymard B (2002) Внутривенный иммуноглобулин при дисфагии миозита с тельцами включения. Неврология 58: 326
Статья. CAS PubMed Google Scholar
Dobloug C, Walle-Hansen R, Gran JT, Molberg Ø (2012) Долгосрочное наблюдение за спорадическим миозитом с тельцами включения, леченным внутривенным иммуноглобулином: ретроспективное исследование с участием 16 пациентов. Clin Exp Rheumatol 30: 838–842
PubMed Google Scholar
Muscle Study Group (2001) Рандомизированное пилотное исследование betaINF1a (Avonex) у пациентов с миозитом с тельцами включения. Неврология 57: 1566–1570
Статья. Google Scholar
Muscle Study Group (2004) Рандомизированное пилотное исследование высоких доз betaINF-1a у пациентов с миозитом с тельцами включения. Неврология 63: 718–720
Статья. CAS Google Scholar
Lindberg C, Trysberg E, Tarkowski A, Oldfors A (2003) Лечение анти-Т-лимфоцитарными глобулинами при миозите с тельцами включения: рандомизированное пилотное исследование. Неврология 61: 260–262
Статья CAS PubMed Google Scholar
Барон Р.Дж., Гербелин Л., Киссель Дж.Т., Кинг В., Маквей А.Л., Саперштейн Д.С., Менделл Дж.Р. (2006) Пилотное испытание этанерцепта при лечении миозита с тельцами включения. Неврология 66: S123 – S124
Статья CAS PubMed Google Scholar
Dalakas MC, Rakocevic G, Schmidt J, et al (2009) Эффект алемтузумаба (CAMPATH 1-H) у пациентов с миозитом с тельцами включения. Мозг 132: 1536–1544
Статья PubMed PubMed Central Google Scholar
Kosmidis ML, Alexopoulos H, Tzioufas AG, Dalakas MC (2013) Эффект анакинры, антагониста рецептора IL1, у пациентов со спорадическим миозитом с тельцами включения (sIBM): небольшое пилотное исследование. J Neurol Sci 334: 123–125
Статья CAS PubMed Google Scholar
Sancricca C, Mora M, Ricci E, Tonali PA, Mantegazza R, Mirabella M (2011) Пилотное испытание симвастатина при лечении спорадического миозита с тельцами включения. Neurol Sci 32: 841–847
Статья PubMed Google Scholar
Hargitai J, Lewis H, Boros I, et al (2003) Бимокломол, соиндуктор белка теплового шока, действует путем длительной активации фактора теплового шока-1. Biochem Biophys Res Commun 307: 689–695
Статья CAS PubMed Google Scholar
Bíró K, Jednákovits A, Kukorelli T, Hegedüs E, Korányi L (1997) Бимокломол (BRLP-42) улучшает периферическую невропатию у крыс с индуцированным стрептозотоцином диабетом. Brain Res Bull 44: 259–263
Статья PubMed Google Scholar
Benatar M, Wuu J, Andersen PM, Atassi N, David W, Cudkowicz M, Schoenfeld D (2018) Рандомизированное двойное слепое плацебо-контролируемое исследование аримокломола в быстро прогрессирующем SOD1 ALS.Неврология 90: e565 – e574
Статья CAS PubMed PubMed Central Google Scholar
Киркегаард Т., Грей Дж., Пристман Д.А. и др. (2016) Терапия на основе белков теплового шока в качестве потенциального кандидата для лечения сфинголипидозов. Sci Transl Med 8: 355ra118–355ra118
Статья CAS PubMed Google Scholar
Амато А.А., Сивакумар К., Гоял Н. и др. (2014) Лечение спорадического миозита с тельцами включения бимагрумабом.Неврология 83: 2239–2246
Статья. CAS PubMed PubMed Central Google Scholar
Lee S-J (2004) Регулирование мышечной массы миостатином. Annu Rev Cell Dev Biol 20: 61–86
Статья CAS PubMed Google Scholar
Mendell JR, Sahenk Z, Al-Zaidy S, et al (2017) Генная терапия фоллистатином при спорадическом миозите с тельцами включения улучшает функциональные результаты.Mol Ther 25: 870–879
Статья CAS PubMed PubMed Central Google Scholar
Greenberg SA (2017) Необоснованные заявления об улучшении функциональных результатов, приписываемых генной терапии фоллистатином при миозите с тельцами включения. Mol Ther 25: 2235–2237
Артикул CAS PubMed PubMed Central Google Scholar
Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM (2002) Пилотное рандомизированное исследование оксандролона при миозите с телец включения.Неврология 58: 1081–1087
Статья. CAS PubMed Google Scholar
Lilleker JB, Bukhari M, Chinoy H (2018) Рапамицин при миозите с тельцами включения: нацеливание на невоспалительные механизмы. Ревматология. DOI: https://doi.org/10.1093/rheumatology/key043
Habers GEA, Takken T (2011) Безопасность и эффективность физических упражнений у пациентов с идиопатической воспалительной миопатией — систематический обзор.Ревматология (Оксфорд) 50: 2113–2124
Статья Google Scholar
Arnardottir S, Alexanderson H, Lundberg IE, Borg K (2003) Спорадический миозит с тельцами включения: пилотное исследование влияния программы домашних упражнений на функцию мышц, гистопатологию и воспалительную реакцию. J Rehabil Med 35: 31–35
Статья PubMed Google Scholar
Johnson LG, Collier KE, Edwards DJ, Philippe DL, Eastwood PR, Walters SE, Thickbroom GW, Mastaglia FL (2009) Улучшение аэробных способностей после программы упражнений при спорадическом миозите с тельцами включения.J Clin Neuromuscul Dis 10: 178–184
Статья PubMed Google Scholar
Spector SA, Lemmer JT, Koffman BM, Fleisher TA, Feuerstein IM, Hurley BF, Dalakas MC (1997) Безопасность и эффективность силовых тренировок у пациентов со спорадическим миозитом с тельцами включения. Muscle Nerve 20: 1242–1248
Статья CAS PubMed Google Scholar
Kwon I, Lee Y, Cosio-Lima LM, Cho J-Y, Yeom D-C (2015) Влияние длительных тренировок с отягощениями на аутофагию в скелетных мышцах крыс спорадического миозита с включениями, вызванного хлорохином.J Exerc Nutr Biochem 19: 225–234
Статья Google Scholar
Oh TH, Brumfield KA, Hoskin TL, Kasperbauer JL, Basford JR (2008) Дисфагия при миозите с тельцами включения. Am J Phys Med Rehabil 87: 883–889
Статья PubMed Google Scholar
Schrey A, Airas L, Jokela M, Pulkkinen J (2017) Ботулинический токсин облегчает дисфагию у пациентов с миозитом с тельцами включения.J Neurol Sci 380: 142–147
Статья CAS PubMed Google Scholar
Claire Langdon P, Mulcahy K, Shepherd KL, Low VH, Mastaglia FL (2012) Глоточная дисфагия при воспалительных заболеваниях мышц, возникающих в результате нарушения надподъязычной мускулатуры. Дисфагия 27: 408–417
Статья PubMed Google Scholar
Карисса Дж. Муньос, Али Х. Маннаа, Дженна Кастеншмидт, Мари Венсель, Намита Гоял, С.Армандо Вильялта, Тахсин Мозаффар (2018) Циркуляционные клетки Kv1.3 + у пациентов с sIBM. Неврология 90. http://n.neurology.org/content/90/15_Supplement/P3.437.
Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJGM, Badrising UA (2011) 12-летнее наблюдение при спорадическом миозите с тельцами включения: конечная стадия с серьезными нарушениями. Brain 134: 3167–3175
Статья PubMed Google Scholar
Benveniste O, Guiguet M, Freebody J, et al (2011) Долгосрочное наблюдательное исследование спорадического миозита с тельцами включения. Brain 134: 3176–3184
Статья PubMed Google Scholar
Дифференциальная диагностика гранулематозной болезни легких: ключи и подводные камни
Реферат
Гранулематозные болезни легких представляют собой гетерогенную группу заболеваний, которые имеют широкий спектр патологий с различными клиническими проявлениями и исходами.Точная клиническая оценка, лабораторные исследования, исследование функции легких, радиологическая визуализация, включая компьютерную томографию с высоким разрешением, а часто и гистопатологическая оценка способствуют достоверной диагностике гранулематозных заболеваний легких. Дифференциальная диагностика является сложной задачей и включает как инфекционные (микобактерии и грибы), так и неинфекционные заболевания легких (саркоидоз, некротизирующий саркоидный гранулематоз, гиперчувствительный пневмонит, горячая ванна легких, бериллиоз, гранулематоз с полиангиитом, эозинофильный гранулематоз, талавангулематоз с полиангиатозом клеточный гистиоцитоз и бронхоцентрический гранулематоз).Бронхоальвеолярный лаваж, эндобронхиальная трансбронхиальная игольчатая аспирация под контролем УЗИ, трансбронхиальная криобиопсия, позитронно-эмиссионная томография и генетическая оценка являются потенциальными кандидатами на повышение точности диагностики гранулематозных заболеваний легких. Поскольку сама по себе гранулема является неспецифическим гистопатологическим признаком, мультидисциплинарный подход важен для уверенного диагноза.
Определение гранулемы
Гранулема — это очаговое скопление воспалительных клеток, активированных макрофагов (эпителиоидных гистиоцитов), гигантских клеток Лангханса и лимфоцитов.Эпителиоидные гистиоциты имеют нечеткие границы клеток и удлиненные ядра, которые отличаются от четко определенных границ клеток и круглых ядер, наблюдаемых в обычных гистиоцитах. Наличие некроза, лимфоцитов, плазматических клеток или многоядерных гигантских клеток не является существенным для образования гранулемы [1]. Казеальный некроз определяется как область в гранулемах с эозинофильными, зернистыми и сыроподобными клеточными остатками с некрозом.
Основная диагностическая процедура и трудности
Дифференциальные диагнозы гранулематозной болезни легких перечислены в таблице 1.Поскольку сама по себе гистологическая аномалия редко является диагностической для конкретного гранулематозного заболевания, диагностическая процедура должна быть сосредоточена на точной клинической оценке, лабораторных исследованиях, обнаружении инфекционных организмов и радиологической оценке. Небольшой размер образцов ткани, полученных с помощью трансбронхиальной биопсии легкого (TBLB), вместе с высокой вариабельностью между наблюдателями среди патологов, усложняет интерпретацию гистопатологии. Хирургическая биопсия легкого может предоставить более крупные образцы ткани по сравнению с TBLB.
ТАБЛИЦА 1Дифференциальная диагностика гранулематозных заболеваний легких
Поскольку инфекция является частой причиной легочных гранулем, всегда важно исключить инфекционные заболевания легких. Наиболее часто в гранулемах легких встречаются микобактерии и грибы. Хотя при инфекционных заболеваниях легких могут наблюдаться как некротизирующие, так и ненекротические гранулемы, некротизирующие гранулемы с большей вероятностью связаны с инфекционными заболеваниями легких [2]. Клиницисты должны учитывать, что при туберкулезе (ТБ) могут также проявляться ненекротические гранулемы, в зависимости от иммунного статуса пациента.
Гистохимические красители, обычно используемые для патологической оценки инфекционных организмов, — это окраска метенамином серебра по Грокотту (GMS) для грибов и окраска по Цилю – Нильсену для микобактерий. Окрашивание периодической кислотой по Шиффу (PAS) также является полезным гистохимическим красителем для грибов. Окраска PAS может обнаруживать клеточные стенки живых грибов, тогда как окраска GMS обнаруживает клеточные стенки как живых, так и мертвых грибковых организмов [3]. Флуоресценция аурамина-родамина показывает большую чувствительность, чем окрашивание по Цилю-Нильсену, хотя специфичность ниже (аурамин-родамин: чувствительность 80% и специфичность 84%; Цил-Нильсен: чувствительность 60% и специфичность 98%) [4].
Следующим шагом после идентификации микобактерий является дифференциация туберкулезных микобактерий от нетуберкулезных микобактерий (НТМ). Однако туберкулезные микобактерии и NTM морфологически схожи и часто неразличимы. В настоящее время единственными окончательными методами дифференциации микобактерий являются микробиологический посев и ПЦР. Микобактерии могут расти в культуре, даже если специфическое окрашивание дает отрицательные результаты. Если микробиологический посев недоступен, ПЦР — единственный метод дифференциации организмов.
Инфекционные болезни легких
Туберкулез
Начало диагностической оценки ТБ обычно основывается на подозрении на ТБ на основании эпидемиологических, клинических и рентгенологических данных. В целом, продолжительный кашель, лимфаденопатия, лихорадка, ночная потливость и потеря веса указывают на туберкулез, но являются неспецифическими. Типичные рентгенологические находки для ТБ включают очаговую инфильтрацию верхней доли (долей), кавитацию, деструкцию тканей, фиброз с тракционными бронхоэктазами, увеличение лимфатических узлов корня / средостения, небольшие узелковые поражения и плевральный выпот.Обнаружение Mycobacterium tuberculosis в мокроте, пробах бронхоскопии, желудочном секрете или плевральной жидкости необходимо для уверенного диагноза. Гранулемы при туберкулезе обычно некротизируются, расположены случайно или бронхиолоцентрически, а также могут поражать кровеносные сосуды [1].
ТБ-ПЦР с использованием эндобронхиальной трансбронхиальной игольной аспирации под контролем УЗИ (EBUS-TBNA) представляет собой новый метод дифференциальной диагностики внутригрудной гранулематозной лимфаденопатии [5].Чувствительность, специфичность, положительная прогностическая ценность, отрицательная прогностическая ценность и диагностическая точность для ТБ составили 56%, 100%, 100%, 81% и 85% соответственно [5]. Неоднородная эхотекстура в EBUS (53% против 13%; p <0,001) и признаки коагуляционного некроза (26% против 3%; p <0,001) в образцах, полученных с помощью EBUS-TBNA, указывают на туберкулез, а не на саркоидоз. [6].
Анализ Xpert MTB / RIF — это новый полуавтоматический тест амплификации полу-вложенных нуклеиновых кислот, который может одновременно идентифицировать M.tuberculosis и устойчивость к рифампицину в течение 2 ч [7–10]. Dhooria et al. [11] исследовал роль Xpert MTB / RIF в сочетании с EBUS-TBNA в дифференцировке ТБ от саркоидоза у 147 пациентов с лимфаденопатией средостения. Xpert MTB / RIF был положительным у 26 (49%) пациентов с ТБ и двух (2%) пациентов с саркоидозом. Xpert MTB / RIF показал хорошую специфичность (98%) и положительную прогностическую ценность (93%) в диагностике ТБ. Соответственно, Xpert MTB / RIF в сочетании с EBUS-TBNA может стать новым инструментом в дополнение к микроскопии мазка для быстрой диагностики у пациентов с подозрением на ТБ.
Нетуберкулезный микобактериоз
NTM — это виды микобактерий, не относящиеся к комплексу M. tuberculosis . На сегодняшний день идентифицировано более 140 видов НТМ. НТМ вызывают поражение самых разных органов, наиболее частыми из которых являются легочные инфекции (65–90%) [12]. Поражения легких при НТМ вызываются в основном комплексом Mycobacterium avium (90%) и Mycobacterium kansasii (10%). Традиционно считалось, что инфекция NTM в легких связана с иммунодефицитом или ранее существовавшим заболеванием легких, таким как хроническая обструктивная болезнь легких или кистозный фиброз.Однако в настоящее время признано, что инфекция NTM в легких также встречается у иммунокомпетентных пациентов без ранее существовавшего заболевания легких [13]. Радиологическое проявление инфильтратов правой средней доли или язычных инфильтратов типично для пожилых женщин, не предрасполагающих к заболеванию легких [14]. У пациентов с ослабленным иммунитетом инфекция NTM характеризуется наличием микобактерий пенистых гистиоцитов, плохо сформированными гранулемами или отсутствием соответствующей воспалительной реакции [15]. Однако у иммунокомпетентных пациентов инфекция NTM демонстрирует широкий спектр гистологических данных, включая воспаление и как некротизирующие, так и ненекротические перибронхиолярные гранулемы [16].Гистологический вид одного только NTM неотличим от туберкулеза. Воздействие NTM в аэрозольной форме может вызвать гиперчувствительное пневмонитоподобное заболевание, известное как «легкое горячей ванны» (как обсуждается в следующем разделе).
Грибковая гранулема
У иммунокомпетентных пациентов воздействие небольшого количества грибка приводит к бессимптомной инфекции. Однако воздействие большого количества грибка может привести к острому гриппоподобному или пневмонийному заболеванию (, например, Coccidioides , Histoplasma и Blastomyces ).Клинические проявления этих инфекций напоминают грипп или внебольничную пневмонию. Диагноз грибковой инфекции ставится преимущественно на основе серологического, а не гистологического исследования [17]. Иногда грибковые организмы сохраняются в хорошо сформированной некротизирующей гранулеме, подобной туберкулезу (, например, Cryptococcus , Coccidioides и Histoplasma ) [18, 19]. Иногда грибковая инфекция прогрессирует, что приводит к хроническому грибковому заболеванию легких. Патологическое проявление этой формы состоит из осложненных некротизирующих гранулем в сочетании с предрасполагающими заболеваниями ( e.грамм. эмфизема и кариес). У пациентов с ослабленным иммунитетом грибковые инфекции могут проявляться в виде диссеминированной формы с плохо сформированными гранулемами и обширными поражениями [20]. Эта форма заболевания часто поражает лимфогематопоэтическую систему и легкие.
Неинфекционные болезни легких
Саркоидоз
Общая диагностическая процедура
Саркоидоз — системная гранулематозная болезнь с гетерогенными клиническими проявлениями. Хотя обычно преобладает поражение легких, могут поражаться и другие органы, и около половины пациентов протекают бессимптомно.Диагноз саркоидоз может быть установлен при выполнении следующих критериев: 1) совместимая клиническая и / или радиологическая аномалия, 2) гистологическое подтверждение неказеозных гранулем и 3) исключение других заболеваний, способных проявлять аналогичные гистологические и клинические проявления [21]. . Типичные гистологические находки саркоидоза — дискретные, хорошо сформированные, интерстициальные, ненекротические эпителиоидно-клеточные гранулемы, показывающие лимфангитное распределение. Инфильтрация лимфоцитов и гранулемы могут быть обнаружены в плевре, межлобулярных перегородках и бронховаскулярных пучках.Хотя неказеозные или некротизирующие гранулемы являются признаком саркоидоза, отдельные области фибриноидного некроза могут быть замечены в центре некоторых гранулем в случаях типичного саркоидоза. Этот тип некроза отличается от казеоза сохранением неповрежденного ретикулинового рисунка, что подтверждается окрашиванием серебром. Частота фибриноидного некроза при саркоидозе колеблется от 6% до 35% [22-25], и может быть связана с выраженными системными симптомами ( например, лихорадка , узловатая эритема и артралгия) и недавним началом саркоидоза.Легочная ткань, за исключением гранулем при саркоидозе, нормальная, в то время как гиперчувствительный пневмонит показывает значительное интерстициальное воспаление даже в областях, кроме гранулем. Гранулематозный васкулит также может наблюдаться при саркоидозе, чего не наблюдается при гиперчувствительном пневмоните [1].
Междисциплинарный подход, включающий клиническую, радиологическую и патологическую оценку, необходим для точного диагноза [26].
Бронхоальвеолярный лаваж
Бронхоальвеолярный лаваж (БАЛ) — один из малоинвазивных и наиболее безопасных исследовательских инструментов для дифференциальной диагностики диффузных паренхиматозных заболеваний легких [27, 28].Характерные признаки БАЛ при саркоидозе включают нормальное или слегка повышенное общее количество клеток с лимфоцитозом, нормальный процент эозинофилов и нейтрофилов, а также отсутствие плазматических клеток и пенистых альвеолярных макрофагов [29, 30]. При активном саркоидозе количество лимфоцитов выше, чем при неактивном саркоидозе. Однако результаты БАЛ могут быть нормальными у 10–15% пациентов, несмотря на активность заболевания. При позднем или запущенном саркоидозе количество нейтрофилов и тучных клеток также может быть увеличено [31].Повышенное количество нейтрофилов в БАЛ может быть связано с неблагоприятным исходом у недавно диагностированных пациентов с саркоидозом [32, 33]. Важность соотношения CD4 + / CD8 + для диагностики саркоидоза является спорной. Отношение CD4 + / CD8 + > 3,5 указывает на присутствие саркоидоза с высокой специфичностью 93–96%, хотя чувствительность низкая, от 53% до 59% [34, 35]. Отношение CD4 + / CD8 + часто высокое у пациентов с синдромом Лёфгрена и острым саркоидозом, тогда как соотношение обычно находится в пределах нормы при неактивном саркоидозе.
Оздемир и др. [36] продемонстрировали, что концентрация CD95 (Fas), апоптотической молекулы в ЖБАЛ (ЖБАЛ), была значительно выше у пациентов с хроническим саркоидозом по сравнению с пациентами со спонтанной ремиссией. Heron et al. [37] оценил полезность интегрина CD103, экспрессированного на CD4 + Т-лимфоцитах в ЖБАЛ для диагностики у 56 пациентов с саркоидозом. Они продемонстрировали, что совместное использование соотношения CD103 + CD4 + / CD4 + (<0.2) с соотношением CD4 + / CD8 + в БАЛ (> 3) или относительным соотношением CD4 + / CD8 + в БАЛ / периферическая кровь (> 2) может отличить саркоидоз от других интерстициальных заболеваний легких. чувствительность 66% и специфичность 89%.
Эндобронхиальная трансбронхиальная игольчатая аспирация под контролем УЗИ
TBLB — традиционная диагностическая процедура для демонстрации гранулемы при легочном саркоидозе с диагностической точностью от 40% до 90% [38-40].Тонкоигольная аспирация под контролем эндоскопической ультрасонографии (EUS-FNA) и EBUS-TBNA являются безопасными и малоинвазивными методами получения гранулематозных образцов [41]. Однородная низкая эхотекстура (88%) и наличие зародышевой центральной структуры (71%) — специфические результаты ультразвукового исследования лимфатических узлов при саркоидозе [42]. Патологические характеристики саркоидоза включают отсутствие некротических остатков или экссудата [43]. В недавнем метаанализе, включающем 14 исследований, диагностическая сила EBUS-TBNA для диагностики саркоидоза была оценена в последовательных популяциях пациентов с внутригрудной лимфаденопатией, независимо от предполагаемой основной этиологии [44].Суммарная диагностическая точность, чувствительность и специфичность составили 79%, 84% и 100% соответственно, что указывает на очень хорошие результаты теста даже в этих неотобранных когортах пациентов с низкой общей распространенностью саркоидоза всего 15%. Однако мало что известно о диагностической точности EBUS-TBNA у пациентов с лимфатическими узлами нормального размера.
Быстрая оценка на месте (ROSE) образцов биопсии, полученных с помощью EBUS-TBNA, может предоставить информацию о количестве лимфатических узлов и проходов, которые необходимо выполнить.Plit et al. [45] продемонстрировали, что EBUS-TBNA с ROSE показала чувствительность, специфичность и положительную прогностическую ценность 88%, 91% и 98%, соответственно, для диагностики саркоидоза. Согласие между наблюдателями между цитотехнологами и патологами было хорошим (κ = 0,91). Недавнее исследование показало, что ROSE более важен для обычного TBNA, чем для EBUS-TBNA; когда обычный TBNA был объединен с ROSE, диагностическая ценность значительно увеличилась и достигла той же чувствительности, что и EBUS-TBNA, тогда как ROSE не увеличивал чувствительность только EBUS-TBNA (TBNA с ROSE 72%, EBUS-TBNA с ROSE 67 %, Только EBUS-TBNA 68% и только TBNA 32%; p = 0.04) [46].
Необходимо учитывать возможные осложнения пневмоторакса и кровотечения [47, 48]. Систематический обзор, включающий 190 исследований с 16 181 пациентом с подозрением на саркоидоз, продемонстрировал, что частота серьезных побочных эффектов составляла 0,14%, а летальности не наблюдалось. Серьезные побочные эффекты были более частыми при использовании EUS-FNA, чем при EBUS-TBNA (0,30% против 0,05%) [49].
Трансбронхиальная криобиопсия
Трансбронхиальная криобиопсия — это новый метод, позволяющий получить образцы паренхимы легких большего размера, чем при обычном TBLB [50].Биопсии получают под рентгеноскопическим контролем с использованием гибкого бронхоскопа, вводимого через жесткую трубку в выбранный бронх. Особое внимание уделяется положению биопсии: криозонд помещают перпендикулярно грудной стенке, чтобы обеспечить точную оценку расстояния от грудной стенки при рентгеноскопии. Оптимальным считается расстояние ∼10 мм от грудной стенки. Зонды охлаждаются углекислым газом, что позволяет температуре на кончике зонда снизиться до -75 ° C в течение нескольких секунд [50].
Tomassetti et al. [51] продемонстрировал повышение диагностической достоверности с добавлением трансбронхиальной криобиопсии в мультидисциплинарную диагностику интерстициальной болезни легких (ILD). Они показали, что трансбронхиальная криобиопсия дала результаты, сопоставимые с хирургической биопсией легкого при диагностике различных ILD. Однако их исследование не оценивало полезность трансбронхиальной криобиопсии для диагностики саркоидоза, потому что только два пациента с окончательным диагнозом саркоидоз были включены в группу, и оба были в группе хирургической биопсии легкого.Ussavarungsi et al. [52] продемонстрировали, что клинические, радиологические и гистопатологические результаты трансбронхиальной криобиопсии дали определенный мультидисциплинарный диагноз у 51% пациентов с ILD (38 из 74 пациентов), включая двух пациентов с саркоидозом. Грифф и др. [53] продемонстрировали, что диагностическая ценность трансбронхиальной криобиопсии составила 83% при саркоидозе (10 из 12 пациентов). Бабяк и др. [54] достиг диагностической эффективности 95% у 41 пациента с ILD, включая шесть с саркоидозом, с помощью этой техники.Основываясь на этих результатах, трансбронхиальная криобиопсия может стать новым диагностическим инструментом саркоидоза.
Позитронно-эмиссионная томография
18 F-фтордезоксиглюкоза (ФДГ) -позитронно-эмиссионная томография (ПЭТ) широко используется при оценке опухолей, васкулитов и воспалительных заболеваний [55] и является более чувствительной, чем сканирование галлия [56, 57]. FDG-PET, вероятно, будет полезен для оценки степени воспалительной активности при саркоидозе в подгруппе пациентов с осложненным течением болезни [58].Mostard et al. [59] показали, что положительное поглощение ПЭТ в легких было связано с тяжестью КТВР и нарушением легочной функции. Vorselaars et al. [60] продемонстрировали, что высокое максимальное стандартизованное значение поглощения легочной паренхимы в FDG-PET на исходном уровне было связано с улучшением форсированной жизненной емкости легких, предполагая, что оценка FDG-PET на исходном уровне полезна для принятия терапевтических решений при саркоидозе. Однако дифференциация гранулематозного воспаления и злокачественного новообразования с помощью FDG-PET / CT все еще остается сложной задачей из-за высокого уровня ложноположительных результатов [61, 62].
l-3- 18 F-фтор-α-метилтирозин (FMT), аналог аминокислоты, накапливается в опухолевых клетках только через систему транспорта аминокислот , что указывает на его более высокую специфичность для оценки злокачественности по сравнению с FDG . Кайра и др. [63] продемонстрировали, что использование FMT-PET в комбинации с FDG-PET может быть полезно для дифференциации саркоидоза от злокачественного новообразования. При раке легких повышенное поглощение на FDG-PET было замечено у 94% пациентов и на FMT-PET у 88% пациентов, тогда как саркоидозные поражения были положительными только на FDG-PET и всегда отрицательными на FMT-PET. 18 F-фтортимидин (FLT) представляет собой новый суррогатный маркер для in vivo оценки синтеза ДНК. FLT использовался для визуализации пролиферации при нескольких злокачественных заболеваниях [64]. Польза FLT-PET при саркоидозе до сих пор остается спорной [65, 66].
Генетика
Генетические факторы могут быть связаны с фенотипом болезни и исходом саркоидоза. Грюневальд и Эклунд [67] продемонстрировали у 150 пациентов с острым началом саркоидоза, что ∼99% пациентов с лейкоцитарным антигеном человека (HLA) -DRB1 * 0301 / DQB1 * 0201 показали спонтанную ремиссию, тогда как только 55% HLA- DRB1 * 0301 / DQB1 * 0201-отрицательные пациенты имели спонтанную ремиссию.Эти аллели кажутся отличными факторами для прогнозирования прогноза при синдроме Лёфгрена. Синдром Лёфгрена и синдром не-Лёфгрена имеют разную генетическую предрасположенность и геномное распределение. Общее перекрытие между этими двумя фенотипами ограничивалось только 17 однонуклеотидными полиморфизмами, включая BTNL2, (бутирофилин-подобный 2) и HLA-DRA [68].
Полиморфизм гена BTNL2 [69–71], HLA-DRB1 * 14 и HLA-DRB1 * 12 [72] являются независимыми факторами риска саркоидоза.Систематический обзор и метаанализ продемонстрировали, что полиморфизм гена BTNL2, G16071A был связан с предрасположенностью к гранулематозной болезни (A против G: OR 1,25; p = 0,005) и особенно к саркоидозу (A против G: OR 1,52; р <0,001) [73]. Генотип D / D ангиотензинпревращающего фермента был связан с повышенным риском саркоидоза (OR 1,21, 95% CI 1,06–1,38; I 2 = 48%) [74]. Тем не менее, в клинике до сих пор нет рутинного применения генетического тестирования для диагностики или дифференциальной диагностики саркоидоза.
Некротизирующий саркоидный гранулематоз
Некротизирующий саркоидный гранулематоз (NSG) — редкое гранулематозное заболевание легких, сопровождающееся васкулитом. До сих пор остается спорным, является ли это дискретным заболеванием или вариантом узлового саркоидоза. Основные особенности NSG включают 1) гистологически саркоидоподобную гранулему с васкулитом и некрозом, 2) рентгенологически множественные легочные узелки без внутригрудной лимфаденопатии и 3) доброкачественное клиническое течение. Клинические симптомы NSG часто неспецифичны ( e.грамм. лихорадка, боль в груди, потеря веса, кашель и одышка) и рентгенологические данные сильно различаются ( например, двусторонних узелков и новообразований, кавитация и плевральный выпот). NSG обычно не влияет на внелегочные органы [75].
Типичные патологические находки включают большие области паренхиматозного некроза, окруженные саркоидоподобным гранулематозным воспалением и гранулематозным васкулитом, непропорциональные гранулематозному воспалению [76]. Некроз при NSG обычно бывает коагулятивным или казеозным.Васкулит почти всегда бывает гранулематозным и затрагивает как вены, так и артерии [75]. Диагноз NSG требует тщательного исключения других подобных заболеваний. Узловой саркоидоз не показывает такого обширного васкулита и диффузного паренхиматозного некроза [3]. Гранулематоз с полиангиитом (ГПА) не связан с саркоидоподобными некротизирующими гранулемами [3]. Гранулематозные инфекции могут быть исключены отрицательными тестами на болезнетворные микроорганизмы. Поскольку невозможно полностью избежать ложноотрицательных микробиологических результатов, несмотря на использование новейших технологий, а гранулематозные инфекции также могут демонстрировать васкулит, некроз и саркоидоподобную реакцию, исключение возможных инфекций особенно важно.
Бронхоцентрический гранулематоз
Бронхоцентрический гранулематоз ограничен легкими и характеризуется деструктивным гранулематозным воспалением бронхиол, которое может быть связано с неспецифической патологической реакцией на различные формы повреждения легких [77]. Заболеваемость и распространенность бронхоцентрического гранулематоза до сих пор неизвестны. Примерно половина всех случаев связана с астмой или аллергическим бронхолегочным аспергиллезом.
В таких случаях типичные результаты включают эозинофилию в крови, повышенный уровень общих сывороточных IgE и антител IgE к Aspergillus видов [78].Окраска по Граму и посев мокроты иногда показывают присутствие Aspergillus или Candida видов. Поскольку бронхоцентрический гранулематоз является частью сложной тканевой реакции на грибковую колонизацию дыхательных путей, также могут наблюдаться другие ассоциированные тканевые проявления гиперчувствительности, включая слизистую закупорку бронхов, эозинофильный бронхиолит и эозинофильную пневмонию.
Хотя рентгенологические данные бронхоцентрического гранулематоза различаются, единичные или множественные легочные узелки и одностороннее уплотнение с преобладанием верхних долей встречаются относительно часто [79].Также могут быть обнаружены массоподобные поражения, альвеолярные инфильтраты и ретикулонодулярные инфильтраты, тогда как внутригрудная лимфаденопатия и кавитация встречаются нечасто [80]. В одном случае ФДГ-ПЭТ продемонстрировал промежуточную активность без значительного поглощения ФДГ бронхоцентрической гранулемой [81].
Бронхоцентрический гранулематоз патологически характеризуется перибронхиолярным некротизирующим гранулематозным воспалением [77]. Преимущественно поражаются бронхиолы по сравнению с более крупными дыхательными путями. Гранулематозное замещение слизистой и подслизистой оболочки палисадными, эпителиоидными и многоядерными гистиоцитами является характерным признаком бронхоцентрического гранулематоза, который приводит к разрушению стенок дыхательных путей [78].Пораженные дыхательные пути могут содержать некротический мусор. При постановке диагноза важен выраженный сопутствующий эозинофильный инфильтрат. Слизистая закупорка бронхов часто встречается в более проксимальных более крупных бронхах. Неспецифическое распространение воспалительного инфильтрата на соседние артерии является частым, но некротизирующего васкулита нет, в отличие от GPA и эозинофильного GPA (EGPA).
Тщательный поиск первопричины, включая аллергический бронхолегочный аспергиллез, микобактериальную и грибковую инфекцию, ревматоидный артрит, GPA и бронхогенную карциному, необходим для точной диагностики бронхоцентрического гранулематоза.
Воспалительное заболевание кишечника
Воспалительные заболевания кишечника (ВЗК) характеризуются ненекротизирующими гранулемами желудочно-кишечного тракта. Хотя внекишечные проявления встречаются у 21–36% пациентов [82], легочные гранулематозные проявления чрезвычайно редки и встречаются менее чем у 1% пациентов [83]. Сообщалось о хроническом бронхиолите с некротизирующими гранулемами без лимфангиитного характера [84]. БАЛ может проявлять лимфоцитоз [27]. Однако одни только патологические и / или микроскопические данные не могут отличить легочное поражение ВЗК от других гранулематозных заболеваний легких.
Гиперчувствительный пневмонит
Общая диагностическая процедура
Гиперчувствительный пневмонит, синоним внешнего аллергического альвеолита, представляет собой сложный синдром, возникающий в результате многократного воздействия различных антигенных частиц, обнаруженных в окружающей среде [85]. Клинические проявления имеют региональные особенности, например. Летний гиперчувствительный пневмонит распространен только в Японии. Частицы, вызывающие гиперчувствительный пневмонит, имеют большое разнообразие размером <5 мкм, включая грибковые ( e.грамм. Aspergillus и Penicillium видов), бактериальные, протозойные, животные (в основном птицы) и насекомые белки, а также низкомолекулярные химические соединения (, например, изоцианатов, цинк, чернила и красители) [86]. Гиперчувствительный пневмонит протекает в острой, подострой или хронической клинической форме, и возможно их наложение. Клиническая картина гиперчувствительного пневмонита зависит от нескольких факторов, включая природу и количество вдыхаемого антигена, интенсивность и частоту воздействия, а также иммунный ответ хозяина, который, вероятно, определяется генетическим фоном [85].
Хотя были предложены различные диагностические критерии гиперчувствительного пневмонита [87, 88], ни один из них не был подтвержден. Lacasse et al. [89] предложил модель клинического прогноза для диагностики гиперчувствительного пневмонита (таблица 2). Если все шесть предикторов в этой модели выполнены, вероятность гиперчувствительного пневмонита составляет 98%. Если ни один из шести предикторов не присутствует, вероятность равна 0%. Гистологическая триада гиперчувствительного пневмонита включает перибронхиолярное хроническое воспаление, плохо сформированные мелкие интерстициальные ненекротизирующие гранулемы и очаги организующейся пневмонии.Многоядерные гигантские клетки случайным образом разбросаны внутри интерстициального воспаления и / или стенок бронхиол.
ТАБЛИЦА 2Предикторы гиперчувствительного пневмонита
Одной из ловушек при диагностике гиперчувствительного пневмонита является эффект курения сигарет. Гиперчувствительный пневмонит у курильщиков встречается реже, чем у некурящих при одинаковом воздействии [90]. Курение сигарет, по-видимому, защищает от развития гиперчувствительного пневмонита. Хотя механизмы защитного действия курения при развитии гиперчувствительного пневмонита плохо изучены, некоторые иммунологические функции, такие как активация макрофагов или пролиферация лимфоцитов, нарушены в легких курильщиков сигарет [88].
Компьютерная томография высокого разрешения
Рентгенография грудной клетки и КТВР обычно являются первыми шагами при обследовании пациента с подозрением на гиперчувствительный пневмонит. Результаты КТВР при остром гиперчувствительном пневмоните могут быть нормальными [91]. У пациентов с более тяжелыми проявлениями острого гиперчувствительного пневмонита HRCT показывает пятнистое или диффузное ослабление матового стекла и / или центрилобулярные плохо определяемые небольшие узелки. Консолидация наблюдается редко (рис. 1a – c) [92–97]. Также наблюдается мозаичная перфузия, которая представляет собой косвенные признаки небольшой обструкции дыхательных путей (воздушной ловушки) из-за сопутствующего бронхиолита.Маленькие узелки неспецифичны для острого гиперчувствительного пневмонита, а также наблюдаются при хроническом гиперчувствительном пневмоните.
РИСУНОК 1Компьютерная томография грудной клетки с высоким разрешением (HRCT) a – c) острого гиперчувствительного пневмонита и d – f) хронического гиперчувствительного пневмонита. КТГ грудной клетки при остром гиперчувствительном пневмоните показывает двустороннюю плотность матового стекла с центрилобулярными микронодулярными акцентом и незначительной консолидацией. КТГ грудной клетки при хроническом гиперчувствительном пневмоните показывает двустороннее ретикулярное затенение, тракционные бронхоэктазы и незначительную мозаичную перфузию вместе с некоторыми микроноузлами.
При подостром гиперчувствительном пневмоните пятнистые воздушные ловушки на снимках выдоха становятся более заметными, часто с дольчатым распределением [93, 98]. Плохо очерченные небольшие узелки более заметны при подостром гиперчувствительном пневмоните по сравнению с острым гиперчувствительным пневмонитом, которые обычно имеют диаметр менее 5 мм и обычно имеют центрилобулярное распределение. Хотя узелки иногда можно увидеть в легких, обычно они распределяются в верхних и средних долях.В результате значительного совпадения подострого и хронического гиперчувствительного пневмонита симптомы хронического гиперчувствительного пневмонита могут наблюдаться при подостром гиперчувствительном пневмоните в различной степени.
При хроническом гиперчувствительном пневмоните наиболее заметными находками на КТВР являются признаки фиброза легких в сочетании с ослаблением матового стекла и центрилобулярными небольшими узелками. Признаки фиброза легких включают утолщение межлобулярной перегородки, потерю долевого объема, линейное / ретикулярное помутнение, тракционные бронхоэктазы и соты (рис. 1d – f) [92, 99–101].Тяговые бронхоэктазы являются важным прогностическим фактором хронического гиперчувствительного пневмонита. Идиопатический легочный фиброз (IPF) можно отличить от хронического гиперчувствительного пневмонита по базальному преобладанию сотов, отсутствию относительной субплевральной щадящей структуры и отсутствию центрилобулярных узелков [99]. Примечательно, что соты наблюдались у 64% пациентов с хроническим гиперчувствительным пневмонитом, что было аналогично частоте, наблюдаемой при IPF [99]. Мозаичная перфузия также может быть полезной для дифференциации IPF от хронического гиперчувствительного пневмонита (отсутствует в IPF, присутствует при гиперчувствительном пневмоните).Неспецифическую интерстициальную пневмонию (NSIP) можно отличить от хронического гиперчувствительного пневмонита по субплевральному сохранению, отсутствию дольчатых областей с матовым стеклом и отсутствию сот [102]. Саркоидоз можно дифференцировать от хронического гиперчувствительного пневмонита по разному распределению микронодулей (перилимфатические / субплевральные / вдоль трещин при саркоидозе против центрилобулярных при гиперчувствительном пневмоните) и отсутствием мозаичной перфузии при саркоидозе [1].
Бронхоальвеолярный лаваж
БАЛ — высокочувствительный метод выявления гиперчувствительного пневмонита. Увеличение общего количества клеток (обычно> 20 × 10 6 в общем 100 мл ЖБАЛ) с заметным увеличением лимфоцитов (обычно> 50%) характерно для гиперчувствительного пневмонита [103]. Лимфоциты ЖБАЛ показывают самое высокое количество при гиперчувствительном пневмоните среди всех ILD. Это увеличение необычно для фиброзных ILD, таких как IPF [104, 105], и лимфоцитоз BAL с пороговым уровнем 30% благоприятно отличает хронический гиперчувствительный пневмонит от IPF [104].Увеличение CD8 + Т-клеток в ЖБАЛ у пациентов с гиперчувствительным пневмонитом приводит к низкому соотношению CD4 + / CD8 + со средними значениями 0,5–1,5. Однако это соотношение варьирует и может часто увеличиваться при хроническом гиперчувствительном пневмоните. Небольшое количество нейтрофилов, эозинофилов, тучных клеток и, что более характерно, плазматических клеток также обнаруживается в ЖБАЛ [95, 106–109].
Активированные Т-клетки при гиперчувствительном пневмоните демонстрируют складчатые ядра и / или широкую цитоплазму и повышенную экспрессию контрлиганда CD28 [110].Макрофаги также активируются при гиперчувствительном пневмоните, показывая пенистые макрофаги и повышенную экспрессию CD80 / CD86 [111]. Подмножества лимфоцитов HLA-DR + CD8 + Т-клеток и естественных Т-клеток-киллеров в ЖБАЛ могут дифференцировать гиперчувствительный пневмонит от саркоидоза. Природные Т-клетки-киллеры были более чем в семь раз выше, а Т-клетки HLA-DR + CD8 + были в два раза выше при гиперчувствительном пневмоните по сравнению с саркоидозом [112].
Цитокины в ЖБАЛ также различаются при гиперчувствительном пневмоните.CCL18 является членом семейства хемокинов CC и обладает хемотаксическим действием для Т-лимфоцитов. Концентрация CCL18 как в сыворотке, так и в ЖБАЛ была значительно увеличена при гиперчувствительном пневмоните по сравнению с IPF, респираторным бронхиолитом, ILD / десквамативной интерстициальной пневмонией и криптогенной организующей пневмонией [113]. Полиморфизм интерлейкина-6 на лиганде хемокинового мотива CXC CXCL5 (ENA78) в ЖБАЛ был специфическим для гиперчувствительного пневмонита по сравнению с саркоидозом и IPF [114].
Трансбронхиальная криобиопсия
Tomassetti et al. [51] продемонстрировал, что 17% пациентов, в основном с идиопатическим NSIP и гиперчувствительным пневмонитом, были переклассифицированы как IPF после получения гистопатологической информации из образцов трансбронхиальной криобиопсии. Ussavarungsi et al. [52] исследовал диагностическую ценность трансбронхиальной криобиопсии у 74 пациентов с диффузным паренхиматозным заболеванием легких. Они отметили, что наиболее частой гистопатологической картиной было гранулематозное воспаление (n = 12, 16%), что привело к окончательному диагнозу гиперчувствительного пневмонита у шести пациентов (8%).Частота пневмоторакса и кровотечения составила 1,4% и 22% соответственно. Грифф и др. [53] продемонстрировали среди 52 пациентов с ILD, что трансбронхиальная криобиопсия была диагностической у шести из семи пациентов (86%) с гиперчувствительным пневмонитом. Несмотря на обнадеживающие результаты трансбронхиальной криобиопсии, использование этого метода пока не рекомендуется в качестве стандартной процедуры для диагностики пневмонита с подозрением на гиперчувствительность, поскольку хронический пневмонит гиперчувствительности и IPF могут быть гистологически похожими, особенно на поздних стадиях.В целом гистологические изменения при хроническом гиперчувствительном пневмоните могут не отличаться от паттернов, обнаруживаемых при других фиброзных заболеваниях легких. При хроническом гиперчувствительном пневмоните сообщалось об изолированных обычных интерстициальных пневмоноподобных или фиброзных паттернах, подобных NSIP. Тем не менее, больший размер образца, полученный с помощью трансбронхиальной криобиопсии, может позволить более уверенно оценить гранулемы и / или другие характерные гистопатологические особенности.
Genetics
Гиперчувствительный пневмонит развивается только у небольшой части людей, подвергшихся воздействию патогенетических антигенов, что позволяет предположить, что дополнительные факторы хозяина / окружающей среды могут играть роль.Falfán-Valenciae et al. [115] исследовал генетическую предрасположенность к гиперчувствительному пневмониту и обнаружил, что частота HLA-DRB1 * 04: 07-DQB1 * 03: 02, DRB1 * 04: 05-DQB1 * 03: 02 и DRB1 * 04: 03- Гаплотипы DQB1 * 03: 02 были выше у пациентов с гиперчувствительным пневмонитом по сравнению со здоровым контролем. Кроме того, комбинация аллелей HLA-DRB1 * 04 и генотипа фактора некроза опухоли-238 GG была значительно увеличена в группе гиперчувствительного пневмонита по сравнению со здоровым контролем (OR 6.93; р = 0,01). Гены низкомолекулярной протеасомы (синоним протеасомной субъединицы β ( PSMB )) кодируют субъединицы фермента, который расщепляет белки на пептиды для основного пути класса I комплекса гистосовместимости. Camarena et al. [116] продемонстрировал значительное увеличение частоты генотипа PSMB8 KQ у пациентов с гиперчувствительным пневмонитом по сравнению со здоровым контролем (OR 7,25, 95% ДИ 2,61–21,3; p = 0,000034). Хотя эти данные свидетельствуют о том, что различные генотипы могут повышать риск развития гиперчувствительного пневмонита, в настоящее время нет клинической ценности генетического тестирования для диагностики или дифференциальной диагностики гиперчувствительного пневмонита.
Гранулематозное заболевание легких, вызванное лекарственными препаратами
Инфекционные заболевания легких, вызванные лекарственными средствами, могут проявляться как гранулематозное заболевание легких с внутригрудной и / или средостенной лимфаденопатией или без нее. Обновленную информацию о токсичности лекарств можно найти на сайте www.pneumotox.com. Гранулемы вызванных лекарственными препаратами ILD, как правило, не вызывают некротизирования. В зависимости от иммунного статуса пациентов необходимы специальные окрашивания и молекулярные анализы, чтобы отличить индуцированные лекарственными средствами ИЛЗ от микобактерий, Pneumocystis или других инфекций.Ряд лекарств (метотрексат, интерферон, Bacillus Calmette-Guérin, инфликсимаб, этанерцепт, лефлуномид, месаламин и сиролимус) могут вызывать гранулемы при респираторных заболеваниях, вызванных лекарственными препаратами. Сама по себе патологическая оценка не позволяет диагностировать гранулематозное заболевание легких, вызванное лекарственными препаратами, поскольку информация о воздействии лекарственных препаратов, вызывающих заболевание, необходима для точного диагноза.
Легкое в гидромассажной ванне
Легкое в гидромассажной ванне — это синдром, сочетающий в себе признаки гиперчувствительного пневмонита и M.avium сложная инфекция, возникающая в результате воздействия загрязненных горячих ванн, спа и джакузи. КТГ грудной клетки показывает ослабление матового стекла и рассеянные небольшие узелки. Патологические данные аналогичны таковым при типичном гиперчувствительном пневмоните. Однако интерстициальная пневмония имеет тенденцию быть менее выраженной, и обычно хорошо сформированы некротизирующие гранулемы, которые часто распределяются в просвете дыхательных путей, а не в перибронхиолярном интерстиции [117]. Для диагностики необходимы культуры и история воздействия.
Бериллиоз
Бериллиоз характеризуется гранулематозной реакцией в легких на вдыхаемый бериллий. Клинические, радиологические и гистопатологические данные имитируют саркоидоз с лимфангиитическим распределением и вовлечением внутригрудных лимфатических узлов [1]. Помимо гранулем, гистопатология выявляет интерстициальное воспаление, которое скорее напоминает гиперчувствительный пневмонит. Наличие в анамнезе воздействия бериллия и положительный результат теста на трансформацию лимфоцитов бериллия имеют решающее значение для точной диагностики бериллиоза.
Тальк-индуцированный гранулематоз
У лиц, злоупотребляющих наркотиками, особенно кокаином и крэком, может развиться широкий спектр острых и хронических заболеваний легких. Способ введения (пероральный, назальный или внутривенный), размер дозы, частота воздействия и наличие связанных веществ связаны с различными легочными проявлениями. Тальк (гидратированный силикат магния) является наиболее часто используемым веществом-носителем для пероральных лекарств. Если такие лекарства вводятся внутривенно наркоманами, типичными патологическими находками являются периваскулярные локализации гранулем, содержащих инородные тела.Тальк содержится в гранулемах в виде пластинчатого вещества [118].
Гранулематоз с полиангиитом
GPA, ранее известный как гранулематоз Вегенера, и микроскопический полиангиит (MPA) определяются как системный васкулит, обычно без эозинофилии, который преимущественно поражает мелкие сосуды, и в настоящее время классифицируются вместе с EGPA в группе, называемой антинейтрофильными цитоплазматическими антителами (антинейтрофильные цитоплазмы). ANCA) -ассоциированный васкулит (AAV) [119]. Положительная доля протеиназы 3 (PRTN3) -ANCA составляет> 90% при расширенном GPA и 75% при ограниченном GPA без поражения почек [120].Типичными гистологическими находками для GPA являются некротизирующие гранулемы, сопровождающиеся некротизирующим васкулитом, напоминающие абсцессы при малом увеличении.
Cabral et al. [121] исследовали клинические характеристики педиатрических пациентов с AAV в когортном исследовании ARChiVe (Регистр детского васкулита: электронная запись). Пожилой возраст начала заболевания (14 против 11 лет), более частые легочные проявления (74% против 44%), менее частые желудочно-кишечные проявления (36% против 58%) и менее частая почечная недостаточность, требующая диализа (13 % против 25%) сформировали характерный клинический профиль для пациентов с GPA по сравнению с пациентами с MPA.Хотя диагностическая ценность FDG-PET / CT для GPA ограничена, FDG-PET / CT может быть полезен для обнаружения скрытых участков активности болезни и степени активности болезни [122].
Меркель и др. [123] идентифицировал аллели риска, относящиеся к AAV, в исследовании ассоциации всего генома и последующей репликации с участием 1986 пациентов с AAV. Они обнаружили, что варианты HLA-DPB1 и HLA-DPA1 были связаны с GPA, а варианты HLA-DQA2 и HLA-DQB1 были связаны с MPA. PRTN3 и SERPINA1 (член 1 семейства серпинов) локусы также были связаны с GPA.
Эозинофильный гранулематоз с полиангиитом
EGPA, ранее известный как синдром Черга-Стросса, определяется как некротизирующее гранулематозное воспаление с выраженной инфильтрацией эозинофилов в дыхательных путях, с некротизирующим васкулитом, преимущественно поражающим сосуды малого и среднего размера и связанным с астма и эозинофилия [119]. Астма и эозинофилия> 1.5 × 10 9 L –1 или 10% лейкоцитов можно обнаружить у всех пациентов с EGPA [124]. Патологическое обследование способствует диагностике EGPA у 57% пациентов, демонстрирующих некротизирующий васкулит сосудов малого и среднего размера (18%), лейкоцитокластический капиллярит (13%), эозинофильную инфильтрацию артериальной стенки (8%) или прилегающие ткани (18%), внесосудистые гранулемы (6%) и / или гигантские клетки (4%). Преобладающие особенности КТВР включают помутнение матового стекла (39%), утолщение бронхиальной стенки (32%), уплотнение (28%) и микронузлы (<3 мм) (24%).Дифференциалы БАЛ показывают среднюю эозинофилию 33%. ANCA положительны на момент постановки диагноза у 31% пациентов [124].
Рецептор 3B Fcγ (FCGR3B) в основном экспрессируется на нейтрофилах и способствует очищению иммунных комплексов нейтрофилами. Марторана и др. [125] продемонстрировали, что дефицит FCGR3B предрасполагает к EGPA и особенно связан с васкулитом при биопсии (OR 3,23, 95% CI 1,3–8,02; p = 0,008).
Ревматоидные узелки
Ревматоидные узелки представляют собой некротизирующие гранулемы, наблюдаемые у 20% пациентов с ревматоидным артритом.Обычно они локализуются подкожно, но могут также встречаться в легких в виде субплевральных некробиотических узелков, множественных или одиночных, с частотой <1% [126]. Размер этих узелков колеблется от 1 до 10 мм. Типичные патологические находки включают обильный центральный некроз с ободком из палисадных гистиоцитов, окруженных инфильтратами лимфоцитов и плазматических клеток. Хотя васкулит может быть обнаружен, некротический васкулит отсутствует [127].
Легочный гистиоцитоз из клеток Лангерганса
Легочный гистиоцитоз из клеток Лангерганса (PLCH), синонимичный эозинофильной гранулеме, является редким заболеванием легких неизвестной причины, в основном поражающим людей молодого возраста [128–130].Поскольку почти все пациенты с ЛКГ у взрослых в прошлом курили сигареты, курение, по-видимому, является одним из важных этиологических факторов. Клинические проявления PLCH обычно неспецифичны, и симптомы включают непродуктивный кашель, одышку, усталость, боль в груди, потерю веса и лихорадку. Однако у некоторых пациентов с ПЛКГ наблюдается опасная для жизни полиорганная недостаточность. КТГ грудной клетки обычно показывает множественные кисты и узелки с преобладанием средних и верхних долей. Эти кисты могут быть изолированными или сливными, иногда имитируя центрилобулярную эмфизему [128].Узелки обычно плохо очерченные или звездчатые, размером 2–10 мм [131, 132].
PLCH патологически характеризуется накоплением активированных клеток Лангерганса в гранулемах, сопровождаемых эозинофилами и лимфоцитами. Гранулемы PLCH связаны с образованием кистозных структур размером более 1 см. Анализ ЖБАЛ обычно показывает присутствие клеток CD1a + и / или CD207 (Лангерин) + (клетки Лангерганса), составляющих> 5% от общего числа клеток [105].Однако пороговое значение 5% окончательно не установлено, и <5% клеток Лангерганса не исключает диагноз PLCH. Низкую долю клеток Лангерганса можно увидеть в других клинических условиях, в том числе у курильщиков, других ИЛЗ и бронхиолоальвеолярной карциномы. Диагностическая ценность TBLB ограничена и составляет всего от 10% до 40% из-за очагового распределения поражений [133].
Сканирование FDG-PET показывает повышенное поглощение PLCH гранулемами. Положительное поглощение ФДГ с большей вероятностью связано с узловой рентгенографической картиной, тогда как отрицательное поглощение ФДГ с большей вероятностью связано с кистозной картиной с меньшим количеством узелков [134].Оберт и др. [135] недавно продемонстрировал, что FDG-PET может иметь некоторую полезность для оценки внелегочных поражений PLCH (, например, костей и щитовидной железы). Однако чувствительность ФДГ-ПЭТ к поражению легких была низкой (трое из 12 пациентов (25%)). Соответственно, роль FDG-PET в диагностике PLCH в настоящее время ограничена.
Гранулематозно-лимфоцитарная интерстициальная болезнь легких
Общий вариабельный иммунодефицит (ОВИН) — это первичный иммунодефицит, характеризующийся дисфункцией В-лимфоцитов и гипогаммаглобулинемией.У пациентов с ОВИН часто наблюдаются рецидивирующие инфекции дыхательных путей [136]. Гранулематозное и лимфопролиферативное воспаление иногда поражает мелкие дыхательные пути и легочный интерстиций, это называется гранулематозно-лимфоцитарным ILD (GLILD). Патологические изменения сложны и включают фолликулярный бронхиолит, лимфоидную гиперплазию, лимфоцитарную интерстициальную пневмонию и саркоидоподобные гранулематозные реакции. Частота ГЛИЛЗ при ОВИН колеблется от 8% до 22% [137]. Нарушение функции Т-клеток и последующее нарушение работы с антигенами рассматривается как возможный механизм GLILD [138].
Общие физические признаки GLILD включают одышку, спленомегалию, лимфаденопатию и / или заболевание печени в контексте мультисистемного гранулематозного / воспалительного поражения. Функциональные тесты легких показывают ограничительную картину с низкой диффузионной способностью легких по монооксиду углерода. При диагностике GLILD необходимо бронхоскопическое исследование на бактериальные и микобактериальные культуры. Нет единого мнения по другим тестам, включая TBLB, ПЦР на микобактерии, другие атипичные патогены, включая вирусы, стандартные тесты на Pneumocystis , дифференциацию клеток ЖБАЛ и фенотипирование лимфоцитов [137].
Типичные радиологические находки GLILD при КТВР включают твердые узелки (<3 см), полутвердые узелки, помутнения в виде чистого матового стекла, пятнистые уплотнения, ретикулярную плотность, увеличенные прикорневые и / или средостенные лимфатические узлы и спленомегалию [137]. FDG-PET / CT может быть полезным для обнаружения областей повышенной метаболической активности в легких и лимфатических узлах, которые могут быть обнаружены с помощью FDG-PET / CT, даже если лимфатические узлы не увеличены [139].
Рекомендуются патологические исследования, включая иммуноокрашивание на CD3, CD4, CD8 и CD20, а также оценку клональности для исключения лимфомы, поскольку существует повышенный риск развития злокачественной лимфомы у пациентов с ОВИН [137].Наличие гранулематозного воспаления, пролиферации перибронхиолярных лимфоидов, интерстициальной лимфоидной пролиферации и преобладания клеток CD4 + указывает на наличие GLILD. Напротив, наличие эозинофилов не характерно для GLILD.
Аспирационная пневмония
Аспирация различных веществ, включая ротоглоточные бактерии, инородные тела и содержимое желудка, приводит к аспирационной пневмонии. Типичные патологические находки включают острую некротизирующую бронхопневмонию, сопровождающуюся гранулемами инородного тела или многоядерными гигантскими клетками, содержащими аспирированный инородный материал [140].Также может быть обнаружена организующаяся пневмония. Не сообщалось о последних достижениях в повышении точности диагностики аспирационной пневмонии.
Тип-специфическая транскриптомика гипоталамических энергочувствительных нейронов, реагирующих на потерю веса
Анализ обогащения аннотации генов (Таблица 1) выборочно выделил в нейронах AGRP транскрипционный ответ для генов, связанных с ER-стрессом: ответ развернутого белка (UPR) и ER-ассоциированная деградация (ERAD) неправильно свернутых белков.Этот путь ранее не исследовался в нейронах AGRP; вместо этого, на основе анализа всего гипоталамуса, UPR был в первую очередь связан с состояниями переедания и резистентностью к лептину (Ozcan et al., 2009), в отличие от состояния дефицита энергии, рассматриваемого здесь. ER-стрессовые ответы возникают во время высоких уровней трансляции белков, когда развернутые белки вызывают программу последующих транскрипционных ответов для увеличения сворачивания и процессинга белка в ER, и этот путь показал наиболее заметное влияние на экспрессию генов, которое мы идентифицировали в нейронах AGRP.Этот паттерн дифференциальной экспрессии генов не наблюдался в нейронах POMC, что указывает на то, что это не артефакт процедуры изоляции нейронов. Следовательно, мы исследовали множество аспектов передачи сигналов ER-стресса путем анализа наших данных RNA-Seq в сочетании с предварительной оценкой передачи сигналов ER-стресса с использованием иммуногистохимии, репортера пути UPR, специфичного для определенного типа клеток, и smFISH.
ER-локализованных белков были значительно превышены в группе генов, регулируемых лишением пищи, в нейронах AGRP (p = 8.3e -16 , гипергеометрический тест), и 95% этих DEG были активированы (фигура 2A). Ключевой маркер UPR, Hspa5 , который кодирует канонический ER-локализованный развернутый чувствительный к белку шаперон BiP, избирательно активируется в нейронах AGRP во время голодания (AGRP: + 3,3 раза, q = 2,5e −10 ; POMC : −1,2 раза, q = 0,52). Соответственно, BiP-иммунореактивность значительно увеличивалась из-за лишения пищи в нейронах AGRP (p = 2,5e -42 , ks-тест, фиг. 2B, D), что указывает на активацию UPR в нейронах AGRP.Напротив, мы также обнаружили, что BiP-иммунореактивность была значительно снижена в нейронах POMC во время голодания (p = 2,5e −19 , ks-тест, рис. 2C, D), что свидетельствует о снижении трансляционной нагрузки белка в этой популяции. .
Недостаток пищи вызывает ответ развернутого белка в нейронах AGRP.
( A ) Log2 (кратное изменение) [log2 (fc)] и q-значения для генов, связанных с локализацией эндоплазматического ретикулума (ER) (путь KEGG: mmu04141), на которые влияет голодание пищи в AGRP (левые столбцы) или нейроны POMC (правые столбцы).( B , C ) Типичные изображения, показывающие BiP-иммунофлуоресценцию от мышей Npy hrGFP или Pomc topazFP . Стрелки: примеры нейронов с флуоресцентной меткой ( B ) AGRP и ( C ) POMC, используемых для количественной оценки BiP. Масштаб: 10 мкм. ( D ) Подсчет популяции соматической интенсивности BiP в нейронах AGRP или POMC. AGRP.fed, n = 209; АГРП.ФД, n = 283; POMC.fed, n = 121; POMC.FD, n = 92; 3 мыши на условие.Столбики: средние значения. Ранговый тест. *** р <0,001. ( E ) Фракция сплайсинга до изоформ транскрипта Xbp1 в нейронах AGRP. Непарный односторонний t-критерий. * р <0,05. ( F , G ) Репрезентативные изображения ( F ) и кумулятивное распределение вероятностей ( G ) TDP43-иммунореактивных гранул (красный) в GFP-экспрессирующих нейронах AGRP (p = 0,76, ks-тест). Fed, n = 276; FD, n = 174; 2 мыши на условие. Масштаб 10 мкм. ( H — J ) Типичные изображения ( H , I ) GFP: выражение ATF6 Agrp Cre ; ai9 (tdtomato) мышей.Кумулятивное распределение вероятностей ( J ) отношения ядерной и цитоплазматической флуоресценции GFP в нейронах AGRP: питание против FD (p = 0,44, ks-тест) Fed, n = 92; FD, n = 63; 4 мыши на условие; или нейроны AGRP: ДМСО против туникамицина (p = 2,9e -16 , ks-тест), ДМСО, n = 42; Туникамицин, n = 38; 1 мышь на условие. ( K ) Дифференциально экспрессируемые гены выживания клеток. Слева средний уровень экспрессии [log2 (TPM)] каждого транскрипта каждой экспериментальной группы. Справа, log2 (кратное изменение) и q-значения для дифференциальной экспрессии между состояниями FD и питания отдельно для нейронов AGRP и POMC.( L ) Схема для DEG в связанных со стрессом путях ER в нейронах AGRP после голодания. Красный: экспрессия активирована, синяя: экспрессия подавлена.
https://doi.org/10.7554/eLife.09800.007UPR включает программу экспрессии гена, которая регулируется Ern1 / Ire1 , которая избирательно увеличивалась в нейронах AGRP при голодании (+2,7 раза, q = 0,001). В ответ на накопление развернутых белков в просвете ER, Ern1 / Ire1 претерпевает конформационное изменение, которое активирует активность эндорибонуклеазы для короткоживущего транскрипта Xbp1 и сплайсирует его в более стабильную мРНК, Xbp1s .Эти данные RNA-Seq показывают повышенное содержание мРНК Xbp1s , сплайсированных в нейронах AGRP после голодания ( Xbp1s / Xbp1 , после кормления: 7,4 ± 1,0%, FD: 15,6 ± 3,8%, p = 0,043, один t-тест; рис. 2E), что соответствует активации эндонуклеазной активности Ern1 / Ire1 в нейронах AGRP после 24-часового голодания.
Xbp1s представляет собой фактор транскрипции, который регулирует экспрессию генов, связанных с UPR. Две хорошо известные Xbp1s-зависимые гены-мишени (Lee et al., 2003) были выборочно активированы в нейронах AGRP после голодания: Dnajc3 / p58 IPK (+ 1,9 раза, q = 0,0007) и Dnajb9 (+ 2,2 раза, q = 0,0003). Многие другие гены, регулируемые Xbp1s, также были увеличены в нейронах AGRP во время голодания, включая шапероны сворачивания белков семейств Hsp40 (Dnaj), Hsp70 (Hspa) и Hsp90, Calr и Canx , а также дисульфид белка. изомеразы ( Pdia3-6 ), которые способствуют укладке белков, катализируя образование дисульфидных связей (рис. 2А). Атм (-3,1 раза, q = 7e -5 ), фермент, воспринимающий повреждение ДНК, который при уменьшении приводит к увеличению сплайсинга Xbp1 (He et al., 2009), подавляется из-за отсутствия пищи. Взятые вместе, Xbp1-сплайсинг и паттерны экспрессии нижележащих генов указывают на ранее не сообщавшуюся роль передачи сигналов Ern1 / Ire1 → Xbp1s в адаптивном ответе нейронов AGRP на дефицит энергии.
Мы также исследовали участие двух других путей передачи сигналов UPR, регулируемых ER-связанными трансмембранными белками, Eif2ak3 (также называемым PERK) и Atf6.Плечо Eif2ak3 / PERK (AGRP: -2,1 раза, q = 0,23) пути UPR первым задействуется во время стресса ER и подавляет трансляцию большей части мРНК. Мы ожидали, что длительная остановка трансляции в нейронах AGRP была маловероятной, поскольку она несовместима с повышенной продукцией нейропептидов в нейронах AGRP во время дефицита энергии. Активация Eif2ak3 / PERK приводит к секвестрации мРНК в частицы рибонуклеопротеина-мессенджера, которые агрегируются в стрессовые гранулы, содержащие РНК-связывающий белок TDP43 (Colombrita et al., 2009). Для оценки образования стрессовых гранул мы использовали иммуноокрашивание против TDP43. Повышенное образование стрессовых гранул не было обнаружено в нейронах AGRP от мышей FD (p = 0,76, ks-тест, рис. 2F, G), что дает предварительные доказательства того, что опосредованная Eif2ak3 / PERK остановка трансляции не может быть задействована при этом 24-часовом питании. момент депривации. Более того, транскрипты для транслокации белка ER ( Srp68 , Srp72 , Sec61a1, Sec61b1, Sec63, Serp1 / Ramp4 ) и трафика Гольджи ( Sec14l1 , Sec22b , Sec24d ) были активированы в нейронах AGRP. возможно, это указывает на повышенную трансляцию белка и способность к укладке во время дефицита энергии и согласуется с потребностью в усилении пептидергической нейротрансмиссии для функции нейрона AGRP.
Дополнительное сигнальное плечо UPR опосредуется через ER-связанный белок Atf6 (AGRP: + 2,0 раза, q = 0,0016). В ответ на стресс ER, Atf6 подвергается протеолитическому расщеплению и высвобождает N-концевой домен фактора транскрипции, который перемещается в ядро и инициирует экспрессию гена для увеличения сворачивания белка. Чтобы изучить степень ядерной транслокации Atf6 в ответе нейронов AGRP на лишение пищи, мы разработали Cre-рекомбиназа-зависимый N-концевой зеленый флуоресцентный белок (GFP): слияние Atf6 (Samali et al., 2010) репортер расщепления Atf6 для использования в головном мозге (рис. 2H). После экспрессии в нейронах AGRP мы измерили соотношение GFP ядро: цитоплазма, которое было очень низким и не увеличивалось значительно у мышей, получавших FD и ad libitum; в то время как индуктор стресса ER, туникамицин, сильно увеличивал ядерную локализацию GFP в нейронах AGRP (питание против FD: p = 0,44, диметилсульфоксид (DMSO) против туникамицина: p = 2,9e −16 , ks-тест, рис. 2H – J). Низкая ядерная локализация указывает на минимальную активацию ответвления UPR, передающего сигнал Atf6, в нейронах AGRP после 24-часового голодания.В соответствии с этим, транскрипт, избирательно регулируемый с помощью Atf6, Herpud1 (Lee et al., 2003), не был значительно (q = 0,22) повышен в нейронах AGRP от мышей FD. Однако, поскольку многие гены, регулируемые Atf6, также регулируются с помощью Xbp1s, устойчивый ER-стрессовый ответ может поддерживаться без передачи сигналов Atf6 (Yamamoto et al., 2007).
Пути окислительного стресса, связанные с повышенной продукцией белка, также увеличиваются в нейронах AGRP во время голодания. Фактор транскрипции Nfe2l2 (также называемый Nrf2 ) регулирует окислительный стресс и обычно быстро разрушается в цитоплазме, частично за счет ассоциации с Keap1 (Kobayashi et al., 2004), который показывает снижение экспрессии при голодании (-2,2 раза, q = 0,05). Nfe2l2 / Nrf2 усиливает экспрессию ключевых ферментов биосинтеза глутатиона Gclc (+ 2,3 раза, q = 0,0002) и Gclm (+ 1,8 раза, q = 0,002), которые повышены в нейронах AGRP с лишением пищи. Nfe2l2 / Nrf2 увеличивает другие транскрипты, связанные с окислительным стрессом, такие как Mt1 (+ 34 раза, q = 0,004), Mt2 (79 раз, q = 0,004) и Srxn1 (+ 17 раз, q = 1.2e -6 ), которые являются одними из наиболее сильно активируемых транскриптов в нейронах AGRP во время голодания. Также были активированы другие транскрипты окислительного стресса, такие как пероксиредоксин 2, Prdx2 (+ 1,9 раза, p = 0,0004), фактор транскрипции Hif1a (+ 1,8 раза, p = 0,005) и окислительный регуляторный фермент пролилгидроксилаза. ( P4hb , + 2,7 раза, p = 8e −7 ). В совокупности эта группа генов, избирательно регулируемых по типу клеток, согласуется с адаптивным ответом на повышенный окислительный стресс в нейронах AGRP во время дефицита энергии.
UPR также обычно приводит к ERAD развернутых белков. Субъединицы E2 убиквитин конъюгированного фермента ( Ube2j1 , Ube2g2 , Ube2q1, Ube2z, Ube4b ) и лигаза E3 Park2 были активированы в нейронах AGRP во время голодания (рис. 2A). Os9 , лектин, который распознает неправильно свернутые белки, и Edem3 и Stt3b (рис. 2A), которые являются ферментами, которые маркируют развернутые белки для ERAD (Sato et al., 2012), также были увеличены.Кроме того, Vcp , компонент ретротранслокации полиубиквитинилированных развернутых белков для деградации, был повышен в нейронах AGRP во время дефицита энергии. В совокупности этот паттерн экспрессии генов указывает на вовлечение ERAD во время голодания.
UPR защищает клетки в течение коротких периодов повышенного ER-стресса, но продолжительная активация может привести к апоптозу. Проапоптотический Ddit3 ( Chop ) обычно активируется во время UPR, однако Ddit3 / Chop не изменился в нейронах AGRP после 24-часового голодания (рис. 2K).Вместо этого были активированы различные антиапоптотические транскрипты Bcl2l ( Bcl-xl ), Manf , Mcl1 и ингибитор каспаз Cflar (рис. 2K). Mcl1 Повышенная регуляция согласуется с ранее установленным путем Creb3l2 → Atf5 → Mcl1 (Izumi et al., 2012), каждый член которого активируется при голодании пищи (Figure 2K). Irak2 (-4,7 раза, q = 3,7e -5 ) был сильно подавлен, что согласуется с сообщениями о том, что его снижение защищает от апоптоза (Benosman et al., 2013). Кроме того, индуцированный клеточным стрессом транскрипт Atf3 (+15,5 раза, q = 0,005) также способствует выживанию нейронов (Francis et al., 2004) и увеличивается при голодании. Этот паттерн экспрессии генов является предварительным свидетельством потенциальной роли антиапоптотических путей в нейронах AGRP во время активации в ответ на дефицит энергии.
Взятые вместе, клеточный тип-специфический профиль транскрипции, иммуногистохимия и репортерная конструкция UPR, специфичная для клеточного типа, указывают на избирательное участие путей ER-стресса в нейронах AGRP во время дефицита энергии.Ожидается, что повышенная активность нейронов и повышенная продукция нейропептидов, связанная с голоданием, увеличит трансляционную нагрузку в нейронах AGRP, но не в POMC, а UPR может служить для борьбы с повышенным нейропептидным и синаптическим выходом. Это частично связано с передачей сигналов Xbp1s, а также приводит к индукции экспрессии генов, связанной с ответами на окислительный стресс, ERAD и защитой клеток от апоптотических путей, которые в противном случае могли бы быть связаны с длительным ER-стрессом (Рисунок 2L).
Мутант SOD1 нарушает аксональный транспорт холинацетилтрансферазы и высвобождение ацетилхолина за счет секвестрирования KAP3 | Молекулярная генетика человека
Аннотация
Мутации в гене супероксиддисмутазы 1 ( sod1 ) вызывают семейный боковой амиотрофический склероз (FALS), вероятно, из-за токсических свойств неправильно свернутого мутантного белка SOD1. Здесь мы продемонстрировали, что, начиная с предварительной стадии FALS, неправильно свернутые виды SOD1 специфически связываются с кинезин-ассоциированным белком 3 (KAP3) в вентральном белом веществе SOD1 G93A -трансгенного спинного мозга мыши.KAP3 представляет собой субъединицу кинезина-2, ответственную за связывание с грузами, включая холинацетилтрансферазу (ChAT). Моторные аксоны у мышей SOD1 G93A -Tg также показали снижение транспорта ChAT с предначальной стадии. Используя новую систему моделирования FALS с использованием клеток NG108-15, мы показали, что микротрубочко-зависимое высвобождение ацетилхолина значительно нарушается неправильно свернутыми видами SOD1. Более того, такое нарушение могло быть нормализовано сверхэкспрессией KAP3.KAP3 также был включен в агрегаты SOD1 в случаях FALS у человека. Эти результаты предполагают, что секвестрация KAP3 неправильно свернутыми видами SOD1 и возникающее в результате ингибирование транспорта ChAT играют роль в дисфункции ALS.
ВВЕДЕНИЕ
Боковой амиотрофический склероз (БАС) — это фатальное прогрессирующее нейродегенеративное заболевание, характеризующееся гибелью клеток моторных нейронов в головном и спинном мозге, сопровождающейся быстрой потерей мышечного контроля и, в конечном итоге, полным параличом (1,2).Подавляющее большинство случаев БАС носит спорадический характер, в то время как ~ 10% случаев являются семейными (БАС). Хотя причина спорадического БАС остается неясной, 15–20% пациентов с БАС имеют точечные мутации в цитозольной супероксиддисмутазе Cu 2+ / Zn 2+ (SOD1). SOD1 — это антиоксидантный фермент, повсеместно экспрессирующийся в цитозоле, который превращает супероксид-анион-радикал в перекись водорода. Выявлено более 115 болезнетворных мутаций, затрагивающих все области продукта гена SOD1.
Предыдущие исследования с использованием трансгенных животных моделей, экспрессирующих мутировавший человеческий SOD1, показали, что заболевание вызвано не потерей его дисмутазной активности, а усилением токсических свойств (1). Многие мутантные белки SOD1 имеют тенденцию легко неправильно складываться и образовывать агрегаты, особенно при окислительном стрессе (3,4). Внутриклеточные агрегаты, содержащие SOD1, были специфически обнаружены в пораженных регионах как у пациентов, так и на животных моделях, обладающих sod1 мутациями (5-7).В спинном мозге мутантных мышей SOD1-Tg неправильно свернутые промежуточные соединения мутантных белков SOD1, которые показали снижение растворимости и увеличение размера, были обнаружены до начала заболевания (8,9). Эти результаты предполагают, что неправильно свернутые молекулы, содержащие мутантный SOD1, могут вносить вклад в специфическое для мотонейрона повреждение при БАС.
Транспорт аксонов необходим для множества нейронных функций, включая синтез, высвобождение и рециклинг нейротрансмиттеров (10). Моторные нейроны часто имеют длинные аксоны, которые у человека могут достигать более метра в длину.По этой причине упорядоченная функция аксонального транспортного механизма должна быть особенно важной для поддержания структуры и передачи сигналов на окончаниях моторных аксонов. Процесс транспорта осуществляется двумя основными семействами моторных белков: суперсемейством кинезина для антероградного транспорта и цитоплазимическим динеином для ретроградного транспорта (11). Сообщалось, что нарушение аксонального транспорта является ранним признаком заболеваний двигательных нейронов, включая БАС (12). У мутантных мышей SOD1-Tg антероградный транспорт нейрофиламентов и тубулина частично нарушается задолго до начала заболевания, предположительно из-за ингибирования определенных транспортных систем (13,14).Что касается ретроградного транспорта, мутации в тяжелой цепи динеина приводят к мотор-селективной дисфункции у мышей (15). Более того, мутация в p150 Glued , субъединице динактина (активатора динеина), была идентифицирована в семье, у которой развилось заболевание, специфичное для нижних мотонейронов (16). Эти результаты убедительно указывают на связь между целостностью аксонального транспортного механизма с функцией и выживанием мотонейронов.
Ранее мы изучали внутриклеточную локализацию неправильно свернутых видов SOD1 путем субклеточного фракционирования спинного мозга от мышей SOD1 G93A -Tg и обнаружили значительное накопление неправильно свернутых видов SOD1 в обогащенной аксонами фракции в начале заболевания (17 ).Из этого результата, вместе с предыдущими сообщениями, мы предположили, что неправильно свернутые виды SOD1 могут связываться с белками, необходимыми для аксонального транспорта, и тем самым ингибировать нормальную функцию моторных белков. Чтобы доказать эту гипотезу, мы провели скрининг молекул, связанных с транспортом аксонов, в поисках белков, которые специфически связываются с неправильно свернутыми видами SOD1, и идентифицировали KAP3 как неправильно свернутый белок, взаимодействующий с SOD1. KAP3 является компонентом моторного комплекса кинезин-2, обеспечивающего аксональный транспорт холинацетилтрансферазы (ChAT) у Drosophila (18,19).Используя разработанную нами систему моделирования in vitro FALS, мы показали, что неправильно свернутая SOD1 ингибирует аксональный транспорт, зависимый от высвобождения ацетилхолина (ACh). Более того, такое нарушение высвобождения ACh может быть устранено сверхэкспрессией KAP3. Эти данные убедительно свидетельствуют о том, что секвестирование KAP3 и возникающее в результате ингибирование транспорта ChAT является новым токсическим свойством мутантных белков SOD1, которое неизбежно приводит к дисфункции, специфичной для двигательных нейронов.
РЕЗУЛЬТАТЫ
Неправильно свернутые виды SOD1 накапливаются в вентральном белом веществе спинного мозга у мышей
SOD1 G93A -Tg до начала заболеванияРанее мы показали с помощью анализа субклеточного фракционирования, что значительное количество неправильно свернутых видов SOD1 находится во фракции, обогащенной ядром и некоторыми видами белков цитоскелета (т.е. глиальный фибриллярный кислый белок и нейрофиламент) в спинном мозге мышей SOD1 G93A -Tg до появления характерных двигательных симптомов БАС (17). Для более детального анализа субклеточной локализации неправильно свернутой SOD1 в моторных нейронах спинного мозга SOD1 G93A -Tg мышей мы разделили спинной мозг на четыре подсегмента с помощью лазерной микродиссекции (LMD) (суммировано на рис. 1A) и использовали лизат из каждого сегмента для обнаружения SOD1 с помощью анализа иммуноблоттинга.Поскольку мы наблюдали, что у мышей SOD1, G93A -Tg, которых мы использовали в этом исследовании, заболевание началось в возрасте около 8 месяцев (246,5 ± 5,8 дней) и умерло около 9,5 месяцев (288,5 ± 7,1 дня, среднее значение ± SEM). , n = 14), мы выбрали для сбора образцов мышей в возрасте 4, 7, 8 и 9 месяцев. Как и ожидалось, большинство неправильно свернутых видов SOD1 (высокомолекулярные полосы / мазки, иммунореактивные для SOD1) локализовались в вентральном сером веществе в возрасте 7 месяцев, то есть за 1 месяц до начала заболевания (рис.1B и C). В этом возрасте мы также обнаружили, что значительное количество неправильно свернутых видов SOD1 было локализовано в вентральном белом веществе, это указывает на то, что неправильно свернутые виды SOD1 локализованы в телах клеток двигательных нейронов и их аксонах. Мы далее разделили вентральный сегмент белого вещества на три подсегмента и подтвердили локализацию неправильно свернутых видов SOD1 в сегменте, обогащенном моторными аксонами, на что указывает экспрессия ChAT (Supplementary Material, Fig. S1). Вместе эти результаты предполагают, что значительное количество неправильно свернутых видов SOD1 находится не только в телах клеток двигательных нейронов, но также в моторных аксонах SOD1 G93A -Tg мышей до начала заболевания.
Рисунок 1.
Неправильно свернутые виды SOD1 были обогащены вентральным белым веществом и вентральным серым веществом в спинном мозге мышей SOD1 G93A -Tg до начала заболевания. ( A ) Схематично показана процедура разделения и получения суботрезков спинного мозга. Нефиксированные замороженные срезы спинного мозга L5 окрашивали 0,01% толуидиновым синим и разрезали на четыре сегмента: дорсальное белое вещество (WM-D), дорсальное серое вещество (GM-D), вентральное белое вещество (WM-V) и вентральное серое вещество. (GM-V), как указано.( B ) Присутствие неправильно свернутых видов SOD1 в подсегментах спинного мозга, полученных от мышей SOD1 G93A -Tg в возрасте 4, 7, 8 и 9,5 месяцев и контрольных мышей дикого типа в возрасте 8 месяцев (без Tg). ) мышей методом иммуноблоттинга. Подлинность образца в каждой дорожке указана над изображением. Полосы / мазки с высокой молекулярной массой, иммунореактивные для SOD1, представляют собой неправильно свернутые виды SOD1, указанные звездочкой. Этот неправильно свернутый вид SOD1 появился как в WM-V, так и в GM-V у мышей SOD1 G93A -Tg через 7 месяцев.Тот же блот анализировали на экспрессию ChAT (маркер двигательного нейрона), нейрофиламента M (NF-M; нейрональный маркер), MBP (маркер миелина) для контролей. Актин служил контролем загрузки. ( C ) Иммунореактивность как мономерной (нормально свернутой), так и неправильно свернутой SOD1, которая показана в анализе иммуноблоттинга в (B), была определена количественно. Уровень экспрессии общего SOD1, а также мономерного и неправильно свернутого SOD1 в произвольных единицах относительно экспрессии актина показан в виде гистограммы.
Рисунок 1.
Неправильно свернутые виды SOD1 были обогащены вентральным белым веществом и вентральным серым веществом в спинном мозге SOD1 G93A -Tg мышей до начала заболевания. ( A ) Схематично показана процедура разделения и получения суботрезков спинного мозга. Нефиксированные замороженные срезы спинного мозга L5 окрашивали 0,01% толуидиновым синим и разрезали на четыре сегмента: дорсальное белое вещество (WM-D), дорсальное серое вещество (GM-D), вентральное белое вещество (WM-V) и вентральное серое вещество. (GM-V), как указано.( B ) Присутствие неправильно свернутых видов SOD1 в подсегментах спинного мозга, полученных от мышей SOD1 G93A -Tg в возрасте 4, 7, 8 и 9,5 месяцев и контрольных мышей дикого типа в возрасте 8 месяцев (без Tg). ) мышей методом иммуноблоттинга. Подлинность образца в каждой дорожке указана над изображением. Полосы / мазки с высокой молекулярной массой, иммунореактивные для SOD1, представляют собой неправильно свернутые виды SOD1, указанные звездочкой. Этот неправильно свернутый вид SOD1 появился как в WM-V, так и в GM-V у мышей SOD1 G93A -Tg через 7 месяцев.Тот же блот анализировали на экспрессию ChAT (маркер двигательного нейрона), нейрофиламента M (NF-M; нейрональный маркер), MBP (маркер миелина) для контролей. Актин служил контролем загрузки. ( C ) Иммунореактивность как мономерной (нормально свернутой), так и неправильно свернутой SOD1, которая показана в анализе иммуноблоттинга в (B), была определена количественно. Уровень экспрессии общего SOD1, а также мономерного и неправильно свернутого SOD1 в произвольных единицах относительно экспрессии актина показан в виде гистограммы.
Неправильно свернутые виды SOD1 ассоциируются с KAP3, компонентом кинезина-2
Предыдущие сообщения убедительно свидетельствуют о том, что нарушение аксонального транспортного механизма связано с дегенерацией мотонейронов (12).Локализация неправильно свернутых видов SOD1 в моторных аксонах SOD1 G93A -Tg мышей предполагает, что неправильно свернутые виды SOD1 могут влиять на функцию белков, необходимых для аксонального транспорта. Антероградный и ретроградный аксональный транспорт опосредуется различными белковыми комплексами, которые состоят из большого количества различных белков. Суперсемейство кинезина для антероградного транспорта, например, делится на шесть семейств (кинезин-1, 2, 3, 4, 13 и 14), состоящих из 45 членов у человека и мыши, и они связываются с соответствующими грузами через молекулы-адаптеры в организме человека. в большинстве случаев (11).Чтобы получить больше информации о взаимосвязи между неправильно свернутой SOD1 и отдельными компонентами комплекса молекулярных моторных белков, мы сравнили седиментацию неправильно свернутой SOD1 при центрифугировании в линейном градиенте плотности с седиментацией репрезентативных молекул, которые составляют моторные комплексы в лизате моторных аксонов. Мы спросили, мигрируют ли какие-либо из этих компонентов моторного комплекса совместно с неправильно свернутым SOD1. Гомогенаты получали из вентрального белого вещества SOD1 G93A -Tg мышей в возрасте 8 месяцев (начало заболевания) и SOD1 WT -Tg мышей, экспрессирующих ген sod1 человека дикого типа. (бессимптомный контроль) и подвергали центрифугированию в линейном градиенте плотности по Найкоденцу.Иммунореактивные полосы SOD1, показывающие ожидаемую молекулярную массу (представляющие нормально свернутую SOD1), наблюдались во фракциях №№ 1-7 у мышей обоих генотипов. С другой стороны, неправильно свернутые виды SOD1 были обнаружены во фракциях около No. 16 только у мышей SOD1 G93A -Tg. Затем мы исследовали профили миграции 17 основных компонентов молекулярных моторов из пяти семейств кинезинов, за исключением кинезина-4, который в основном экспрессируется в ювенильном мозге, цитоплазматического динеина и динактина.Среди этих молекул KAP3, субъединица моторного комплекса кинезин-2, явно совместно мигрировала с неправильно свернутыми видами SOD1 вокруг фракции № 16 из SOD1 G93A -Tg мышей (фиг. 2A, звездочки). В дополнение к KAP3, KIF3A и KIF3B, которые являются партнерами по связыванию KAP3, были слабо обнаружены вокруг фракции No. 16. Мы также наблюдали p150 Glued , субъединицу динактина, мигрирующую вместе с неправильно свернутыми видами SOD1. Однако мы пока не приступили к подробному анализу этого наблюдения, поскольку p50, другая субъединица динактина (данные не показаны) или другие компоненты динеина, такие как промежуточная цепь динеина (динеин IC на рис.2А) не мигрировали совместно с неправильно свернутыми видами SOD1. Чтобы подтвердить эту ассоциацию KAP3 с неправильно свернутой SOD1, мы выполнили иммунопреципитационный анализ с использованием фракций №№ 16 и 7. KAP3 был идентифицирован в иммунопреципитатах против SOD1, полученных из фракции № 14, с. 16, содержащие неправильно свернутые формы SOD1, но не во фракции из № 7 (рис. 2Б). При реципрокной иммунопреципитации мы обнаружили неправильно свернутую SOD1 в иммунопреципитатах анти-KAP3 только во фракции No. 16. CRMP-2 (медиаторный белок-2 ответа на коллапсин), аналог KAP3 для моторного комплекса кинезин-1, не ассоциировался с неправильно свернутым SOD1.KAP3, по-видимому, связан с неправильно свернутыми видами SOD1, по крайней мере, за 2 месяца до начала заболевания, поскольку неправильно свернутые виды SOD1 были обнаружены в иммунопреципитате с помощью антитела против KAP3 из экстрактов 6-месячного SOD1 G93A — Tg спинного мозга (рис. 2C). Кроме того, мы исследовали иммуногистохимическую локализацию SOD1 и KAP3 на срезах спинного мозга SOD1 G93A -Tg мышей в возрасте 9 месяцев (конечная стадия). Мы обнаружили, что белковые агрегаты в остальных моторных нейронах спинного мозга, иммунореактивные по отношению к KAP3, также были положительными по SOD1 (рис.2D, верхняя панель). KAP3-положительные агрегаты также содержали иммунореактивность убиквитина, что позволяет предположить, что агрегаты представляют собой клеточные тельца включения (фиг. 2D, нижняя панель). Эти результаты предполагают, что неправильно свернутые виды SOD1 специфически связываются с KAP3 в моторных аксонах SOD1 G93A -Tg мышей и что эта ассоциация начинается до появления моторных симптомов.
Рисунок 2.
Неправильно свернутые виды SOD1 в вентральном белом веществе спинного мозга специфически ассоциируют с KAP3.( A ) Фракции, полученные центрифугированием в градиенте плотности по Nycodenz вентрального белого вещества 8-месячного ребенка SOD1 WT -Tg и SOD1 G93A -Tg спинного мозга, анализировали на наличие указанных белков (SOD1 и отдельные компоненты моторных комплексов кинезина и динеина) методом иммуноблоттинга. Каждая дорожка (слева направо) представляет собой аликвоту, собранную сверху. Обратите внимание, что неправильно свернутые виды SOD1 (обозначенные звездочками) мигрировали вокруг фракции №16 в SOD1 G93A -Tg спинного мозга, куда совместно мигрировал KAP3. ( B ) Иммунопреципитаты антител против SOD1 и KAP3 (обозначенных как «IP: SOD1» или «IP: KAP3») из фракций 7 и 16 центрифугирования в градиенте плотности по Найкоденцу (F7, F16) SOD1 G93A Спинной мозг мыши -Tg, приготовленный выше, анализировали на присутствие указанных белков с помощью иммуноблоттинга. Также показан иммуноблот образцов F7 и F16 (20% от количества, используемого для иммунопреципитации), исследованных на экспрессию того же набора белков.Обратите внимание, что ассоциация неправильно свернутых SOD1-KAP3 была обнаружена из F16, но не из F7. ( C ) Иммунопреципитаты антитела против KAP3 из экстрактов цельного спинного мозга 4-, 6- и 8-месячных мышей SOD1 G93A -Tg мышей и 8-месячных мышей SOD1 Мышей WT -Tg анализировали на наличие неправильно свернутой SOD1 с помощью иммуноблоттинга. KIF3A и KIF3B, которые являются компонентами кинезина-2, служили контролем для иммунопреципитации. Обратите внимание, что неправильно свернутые виды SOD1 (звездочки) были захвачены антителом против KAP3 в 6- и 8-месячных экстрактах SOD1 G93A -Tg.( D ) Репрезентативные фотографии иммуногистохимической совместной локализации KAP3 и SOD1 (верхняя панель) и KAP3 и убиквитина (нижняя панель) у 9-месячного SOD1 G93A -Tg спинного мозга мыши. На вставках показаны изображения выделенной области с большим увеличением. Стрелки указывают на агрегатоподобные структуры в телах клеток двигательных нейронов, которые были положительными как для KAP3, так и для SOD1 (верхняя панель) и KAP3 и убиквитина (нижняя панель). Масштабные линейки = 50 мкм.
Рисунок 2.
Неправильно свернутые виды SOD1 в вентральном белом веществе спинного мозга специфически ассоциируют с KAP3. ( A ) Фракции, полученные центрифугированием в градиенте плотности по Nycodenz вентрального белого вещества 8-месячного ребенка SOD1 WT -Tg и SOD1 G93A -Tg спинного мозга, анализировали на наличие указанных белков (SOD1 и отдельные компоненты моторных комплексов кинезина и динеина) методом иммуноблоттинга.Каждая дорожка (слева направо) представляет собой аликвоту, собранную сверху. Обратите внимание, что неправильно свернутые виды SOD1 (обозначенные звездочками) мигрировали вокруг фракции № 16 в SOD1 G93A -Tg спинного мозга, куда совместно мигрировал KAP3. ( B ) Иммунопреципитаты антител против SOD1 и KAP3 (обозначенных как «IP: SOD1» или «IP: KAP3») из фракций 7 и 16 центрифугирования в градиенте плотности по Найкоденцу (F7, F16) SOD1 G93A Спинной мозг мыши -Tg, приготовленный выше, анализировали на присутствие указанных белков с помощью иммуноблоттинга.Также показан иммуноблот образцов F7 и F16 (20% от количества, используемого для иммунопреципитации), исследованных на экспрессию того же набора белков. Обратите внимание, что ассоциация неправильно свернутых SOD1-KAP3 была обнаружена из F16, но не из F7. ( C ) Иммунопреципитаты антитела против KAP3 из экстрактов цельного спинного мозга 4-, 6- и 8-месячных мышей SOD1 G93A -Tg мышей и 8-месячных мышей SOD1 Мышей WT -Tg анализировали на наличие неправильно свернутой SOD1 с помощью иммуноблоттинга.KIF3A и KIF3B, которые являются компонентами кинезина-2, служили контролем для иммунопреципитации. Обратите внимание, что неправильно свернутые виды SOD1 (звездочки) были захвачены антителом против KAP3 в 6- и 8-месячных экстрактах SOD1 G93A -Tg. ( D ) Репрезентативные фотографии иммуногистохимической совместной локализации KAP3 и SOD1 (верхняя панель) и KAP3 и убиквитина (нижняя панель) у 9-месячного SOD1 G93A -Tg спинного мозга мыши.На вставках показаны изображения выделенной области с большим увеличением. Стрелки указывают на агрегатоподобные структуры в телах клеток двигательных нейронов, которые были положительными как для KAP3, так и для SOD1 (верхняя панель) и KAP3 и убиквитина (нижняя панель). Масштабные линейки = 50 мкм.
Аксональный транспорт ChAT был значительно нарушен в спинномозговых мотонейронах до начала заболевания у мышей SOD1
G93A -TgКинезин-2 представляет собой повсеместно и широко экспрессируемый молекулярно-моторный комплекс, который состоит из KAP3 и KIF3A вместе с KIF3B или KIF3C (20).Моторный домен, состоящий из KIF3A вместе с KIF3B или KIF3C, перемещается по микротрубочкам с гидролизом АТФ, а KAP3 определяет конкретный груз, который необходимо транспортировать. Недавняя работа с использованием ганглиозных клеток Drosophila показала, что кинезин-2 участвует в транспорте ChAT и ACh эстеразы (AChE) (18). Хотя оба фермента необходимы для эффективного снабжения ACh нервными окончаниями мотонейронов спинного мозга, количество поставляемого ChAT более критично для общего производства ACh (21).Следовательно, чтобы изучить влияние ассоциации неправильно свернутых SOD1-KAP3 на функцию моторных нейронов у мышей SOD1 G93A -Tg во время прогрессирования заболевания, мы решили проанализировать транспорт ChAT в моторных нейронах. Чтобы достичь этого, мы получили белковые комплексы мотор-груз, используя ранее описанный метод из вентральных корешков хвостового спинного мозга, которые представляют моторные аксоны, только что отошедшие от спинного мозга, и исследовали присутствие ChAT с помощью анализа иммуноблоттинга.Мы обнаружили, что значительно более низкий уровень ChAT присутствовал в конском хвосте мышей SOD1 G93A -Tg по сравнению с таковыми у мышей SOD1 WT -Tg в возрасте 7 месяцев или позже (рис. 3А). Кроме того, моторные компоненты кинезина-2, KAP3, KIF3A и KIF3B, все показали аналогичное снижение экспрессии во время прогрессирования заболевания конского хвоста мышей SOD1 G93A -Tg. С другой стороны, KHC (тяжелая цепь кинезина), KLC (легкая цепь кинезина) (компоненты кинезина-1) и Rab3 (белок, связанный с синаптическими пузырьками, транспортируемый KIF1A) постоянно экспрессировались во время прогрессирования заболевания.Снижение транспорта ChAT в предсимптоматическом SOD1 G93A -Tg седалищном нерве мыши также было продемонстрировано с помощью анализа лигирования (дополнительный материал, фиг. S2A). Эти результаты предполагают, что количество ChAT, транспортируемое комплексом кинезин-2, значительно снижается в моторных аксонах мышей SOD1 G93A -Tg до начала заболевания.
Рисунок 3.
Аксональный транспорт ChAT был избирательно нарушен в моторных нейронах спинного мозга перед началом заболевания у мышей SOD1 G93A -Tg.( A ) Компоненты транспорта аксонов, полученные из вентральных корешков хвостового спинного мозга мышей SOD1 WT -Tg и SOD1 G93A -Tg мышей указанного возраста анализировали на наличие указанных белков. с помощью иммуноблот-анализа (левая панель). Уровень экспрессии каждой молекулы, исследованной для мышей SOD1 G93A -Tg, показан как процент от уровня в возрасте 4 месяцев (правая панель).Обратите внимание, что снижение уровней обнаружения компонентов ChAT и кинезина-2 (KAP3, KIF3A и KIF3B) у мышей SOD1 G93A -Tg стало очевидным в возрасте 7 месяцев, за 1 месяц до начала заболевания. ( B ) Фракции, полученные центрифугированием в градиенте плотности сахарозы компонентов, транспортируемых через аксоны, от 8-месячных мышей, анализировали на наличие указанных белков (SOD1 и отдельные компоненты моторных комплексов кинезина) с помощью анализа иммуноблоттинга.Каждая дорожка (слева направо) представляет собой аликвоту, собранную сверху. Обратите внимание, что только компоненты кинезина-2 мигрировали вместе с неправильно свернутыми видами SOD1 (обозначены звездочками на блотах).
Рисунок 3.
Аксональный транспорт ChAT был избирательно нарушен в моторных нейронах спинного мозга перед началом заболевания у мышей SOD1 G93A -Tg. ( A ) Компоненты транспорта аксонов, полученные из вентральных корешков хвостового спинного мозга мышей SOD1 WT -Tg и SOD1 G93A -Tg мышей указанного возраста анализировали на наличие указанных белков. с помощью иммуноблот-анализа (левая панель).Уровень экспрессии каждой молекулы, исследованной для мышей SOD1 G93A -Tg, показан как процент от уровня в возрасте 4 месяцев (правая панель). Обратите внимание, что снижение уровней обнаружения компонентов ChAT и кинезина-2 (KAP3, KIF3A и KIF3B) у мышей SOD1 G93A -Tg стало очевидным в возрасте 7 месяцев, за 1 месяц до начала заболевания. ( B ) Фракции, полученные центрифугированием в градиенте плотности сахарозы компонентов, транспортируемых через аксоны, от 8-месячных мышей, анализировали на наличие указанных белков (SOD1 и отдельные компоненты моторных комплексов кинезина) с помощью анализа иммуноблоттинга.Каждая дорожка (слева направо) представляет собой аликвоту, собранную сверху. Обратите внимание, что только компоненты кинезина-2 мигрировали вместе с неправильно свернутыми видами SOD1 (обозначены звездочками на блотах).
Результаты, представленные на фиг. 2A, показывают, что ChAT не мигрирует совместно с неправильно свернутым комплексом SOD1-KAP3, указывая тем самым, что KAP3 не связывается с ChAT и неправильно свернутым SOD1 одновременно. Уменьшение количества ChAT и моторного комплекса кинезин-2 в аксонах мотонейронов у мышей SOD1 G93A -Tg вместе с данными на Рисунке 2A предполагают, что неправильно свернутые виды SOD1 могут ингибировать опосредованный кинезином-2 транспорт ChAT. путем захвата моторного комплекса кинезин-2 в теле клетки двигательного нейрона и в окружающих областях.Для лучшего понимания поведения неправильно свернутых видов SOD1 в моторных аксонах мышей SOD1 G93A -Tg мы сравнили профиль миграции при центрифугировании в градиенте плотности сахарозы моторных компонентов кинезина и ChAT с профилем SOD1, используя комплексы аксональный мотор-груз белков, приготовленные выше в качестве образца. Мы обнаружили, что небольшое количество неправильно свернутых видов SOD1 присутствует в комплексах аксональный мотор-груз-белок и мигрирует вместе с небольшой фракцией KAP3, KIF3A (рис.3B) и KIF3B, хотя для обнаружения KIF3B требуется очень долгая выдержка (данные не показаны). Мы также обнаружили, что ChAT коиммунопреципитирует с KAP3 во фракциях, в которых компоненты KAP3 и кинезин-2 мигрировали совместно. (Дополнительный материал, рис. S2B). Эти результаты предполагают, что неправильно свернутые виды SOD1 не только захватывают моторный комплекс kinesin-2 посредством ассоциации с KAP3 внутри и вокруг клеточных тел, но некоторые из неправильно свернутых SOD1 также транспортируются к аксонам вместо нормальных грузов, таких как ChAT.
Клетки NG108-15 служат моделью
in vitro для FALSМы показали четкую корреляцию между ассоциацией неправильно свернутой SOD1 с KAP3 и ингибированием транспорта ChAT в пределах SOD1 G93A -Tg моторных аксонов мыши.Чтобы детально изучить причинную связь между этими двумя явлениями, мы разработали новую модель клеточной культуры для FALS, в которой клетки проявляют холинергические нейроноподобные свойства: ChAT транспортируется в аксоноподобных процессах, ACh высвобождается путем деполяризации, а мутантный SOD1 — неправильно свернутый / агрегированный. Для этой цели мы использовали клетки NG108-15, которые происходят из нейронов / глии и, как известно, дифференцируются в холинергический нейроноподобный фенотип (22,23). Сначала мы клонировали производную от одной клетки подлинию клеток NG108-15, которую мы назвали PNG3 (очищенная клетка NG108-15-3), которая показала самую высокую экспрессию ChAT при дифференцировке дибутирил-цАМФ и дексаметазоном (данные не показаны).Мы наблюдали, что экспрессия ChAT и синаптофизина (интегральный мембранный белок, обнаруживаемый в синаптических везикулах) достигает максимального уровня примерно через 72 часа после стимуляции (рис. 4A). Путем иммуноцитохимии мы обнаружили, что экспрессия ChAT и KAP3 преимущественно определялась в рамках расширенных процессов PNG3 через 72 часа после стимуляции (фиг. 4B), как и другие субъединицы кинезина-2 (данные не показаны). На основании этих наблюдений мы решили использовать клетки PNG3 в качестве модели холинергических нейронов на 3-й день после начала дифференцировки для всех следующих экспериментов.
Рисунок 4.
КлеткиPNG3 служили в качестве in vitro модельной системы для изучения токсичности мутантного SOD1 на холинергические нейроны. ( A ) Экспрессию указанных белков в клетках PNG3 во время холинергической нейроноподобной дифференцировки исследовали с помощью анализа иммуноблоттинга. Клетки PNG3 лизировали в указанные сроки после индукции дифференцировки дибутирил-цАМФ и дексаметазоном. Показаны репрезентативные изображения иммуноблотов (левая панель) и количественного анализа блотов (правая панель).Уровень экспрессии нормализован по актину и относительно уровня нормального спинного мозга в каждый момент времени для каждой молекулы. ( B ) Показаны репрезентативные фотографии экспрессии ChAT и KAP3 в дифференцированных клеточных телах PNG3 и процессов, обнаруженных с помощью иммуноцитохимии. Масштабная линейка = 25 мкм. ( C ) Генерацию неправильно свернутых видов SOD1 в клетках PNG3, сверхэкспрессирующих FLAG-меченную SOD1, несущую мутацию WT, G93A, G85R или A4V, оценивали с помощью анализа иммуноблоттинга. Трансфицированные клетки PNG3 были либо необработанными, либо обработанными MG132, либо обработанными MG132, а затем H 2 O 2 (как указано выше на изображениях).Одну предварительно растворимую и нерастворимую фракции Triton X-100 анализировали отдельно на SOD1-FLAG, KAP3, KHC и актин (для контроля нагрузки). Спинной мозг мышей SOD1 G93A -Tg в возрасте 2 и 9 месяцев также подвергали тому же анализу для сравнения.
Рисунок 4.
КлеткиPNG3 служили in vitro модельной системой для изучения токсичности мутантного SOD1 на холинергические нейроны. ( A ) Экспрессию указанных белков в клетках PNG3 во время холинергической нейроноподобной дифференцировки исследовали с помощью анализа иммуноблоттинга.Клетки PNG3 лизировали в указанные сроки после индукции дифференцировки дибутирил-цАМФ и дексаметазоном. Показаны репрезентативные изображения иммуноблотов (левая панель) и количественного анализа блотов (правая панель). Уровень экспрессии нормализован по актину и относительно уровня нормального спинного мозга в каждый момент времени для каждой молекулы. ( B ) Показаны репрезентативные фотографии экспрессии ChAT и KAP3 в дифференцированных клеточных телах PNG3 и процессов, обнаруженных с помощью иммуноцитохимии.Масштабная линейка = 25 мкм. ( C ) Генерацию неправильно свернутых видов SOD1 в клетках PNG3, сверхэкспрессирующих FLAG-меченную SOD1, несущую мутацию WT, G93A, G85R или A4V, оценивали с помощью анализа иммуноблоттинга. Трансфицированные клетки PNG3 были либо необработанными, либо обработанными MG132, либо обработанными MG132, а затем H 2 O 2 (как указано выше на изображениях). Одну предварительно растворимую и нерастворимую фракции Triton X-100 анализировали отдельно на SOD1-FLAG, KAP3, KHC и актин (для контроля нагрузки).Спинной мозг мышей SOD1 G93A -Tg в возрасте 2 и 9 месяцев также подвергали тому же анализу для сравнения.
Чтобы использовать клетки PNG3 в качестве модели клеточной линии для FALS, необходимо наблюдать неправильное сворачивание / образование агрегатов за счет сверхэкспрессии мутантного SOD1. Окислительный стресс имеет тенденцию вызывать неправильную укладку мутантных белков SOD1 и формирование агрегатов (4). Экспериментально можно наблюдать образование агрегатов мутантного SOD1, которому способствует ингибирование протеасомной активности (24,25).Следовательно, мы использовали комбинированное состояние повышенного окислительного стресса плюс снижение протеасомной активности для индукции неправильного свертывания SOD1 и для увеличения накопления этих неправильно свернутых видов SOD1 в дифференцированных клетках PNG3, сверхэкспрессирующих мутантный SOD1. Мы трансфицировали клетки PNG3 с помощью ранее хорошо изученных мутаций G93A, G85R и A4V и контроля дикого типа человеческого SOD1 для сверхэкспрессии (3). Неправильно уложенные виды SOD1 были обнаружены в 1% нерастворимых фракциях Triton X-100 лизатов, полученных из всех трех типов FALS-связанных мутантных трансфектантов SOD1, но не в клетках, сверхэкспрессирующих SOD1 дикого типа (рис.4С). Мы также обнаружили, что белок SOD1, несущий мутацию G85R, неправильно уложился без увеличения окислительного стресса, что свидетельствует о ранее сообщавшейся нестабильной природе этого мутантного белка (24). Эти результаты предполагают, что мутантный SOD1 может образовывать неправильно свернутые и / или агрегированные молекулы в дифференцированных клетках PNG3, что очень похоже на то, что наблюдается у мышей SOD1 G93A -Tg при патологических стрессах.
Для анализа патологической значимости ассоциации неправильно свернутой SOD1-KAP3 в этой модельной системе клеток PNG3 мы спросили, наблюдается ли в этой модели ассоциация KAP3 с неправильно свернутой SOD1, как мы видели в SOD1 G93A -Tg мыши.Чтобы напрямую доказать ассоциацию KAP3 с неправильно свернутой SOD1, мы провели эксперименты по иммунопреципитации с использованием лизатов клеток PNG3, трансфицированных SOD1 дикого типа или мутантной SOD1, а затем обработанных перекисью водорода, чтобы вызвать неправильную укладку мутантной SOD1. Мы обнаружили, что иммунопреципитаты анти-KAP3 содержали неправильно свернутые виды SOD1, а антитело против мутантного SOD1-Flag иммунопреципитировало неправильно свернутый SOD1 вместе с KAP3 (фиг. 5A). Чтобы визуализировать ассоциацию неправильно свернутой SOD1-KAP3 в клетках PNG3, мы выполнили иммуноцитохимический анализ локализации KAP3 и мутантной SOD1.Мы обнаружили, что мутантные SOD1-иммунореактивные агрегаты были заметны в клеточных телах, а также в процессах обработанных MG132 дифференцированных клеток PNG3, экспрессирующих G85R SOD1, и эти агрегаты также были положительными для KAP3 (фиг. 5B и C). Эти наблюдения вместе предполагают, что клеточная модель FALS, которую мы разработали здесь с использованием клеток PNG3, воспроизводит ассоциацию KAP3 и неправильно свернутой SOD1, наблюдаемой у SOD1 G93A -Tg мышей и, следовательно, подходит для анализа патологического воздействия неправильно свернутой SOD1 -KAP3 ассоциация на моторных нейронах.
Рисунок 5.
KAP3 ассоциирует с неправильно свернутыми видами SOD1 в дифференцированных клетках PNG3. ( A ) Иммунопреципитаты антител против FLAG и KAP3 (помеченных как «IP: FLAG» или «IP: KAP3») из лизатов клеток PNG3, трансфицированных плазмидами экспрессии для SOD1 или SOD1 дикого типа с помощью G93A, G85R или A4V мутации с индукцией неправильного сворачивания или без нее с помощью MG132 / H. 2 O 2 анализировали на наличие указанных белков с помощью иммуноблот-анализа.Также показан иммуноблот лизатов, используемых для иммунопреципитации (20% количества) экспрессии того же набора белков. Обратите внимание, что ассоциация неправильно свернутых SOD1-KAP3 была обнаружена во всех трех мутантных трансфектантах SOD1 (обозначенных звездочками). ( B и C ) Фотографии KAP3 и FLAG-меченного SOD1 в дифференцированных клетках PNG3, трансфицированных либо FLAG-меченным SOD1 дикого типа, либо SOD1, несущим мутацию G85R до и после обработки MG132, показаны на (B) и ( C) соответственно.Справа от каждого набора также показаны изображения фазового контраста. Звездочки указывают на нетрансфицированные клетки. Обратите внимание, что заметное образование агрегатов происходит только в мутантных SOD1-экспрессирующих клетках и процессах после обработки MG132. Масштабные линейки = 25 мкм.
Рисунок 5.
KAP3 ассоциирует с неправильно свернутыми видами SOD1 в дифференцированных клетках PNG3. ( A ) Иммунопреципитаты антител против FLAG и KAP3 (помеченных как «IP: FLAG» или «IP: KAP3») из лизатов клеток PNG3, трансфицированных плазмидами экспрессии для SOD1 или SOD1 дикого типа с помощью G93A, G85R или A4V мутации с индукцией неправильного сворачивания или без нее с помощью MG132 / H. 2 O 2 анализировали на наличие указанных белков с помощью иммуноблот-анализа.Также показан иммуноблот лизатов, используемых для иммунопреципитации (20% количества) экспрессии того же набора белков. Обратите внимание, что ассоциация неправильно свернутых SOD1-KAP3 была обнаружена во всех трех мутантных трансфектантах SOD1 (обозначенных звездочками). ( B и C ) Фотографии KAP3 и FLAG-меченного SOD1 в дифференцированных клетках PNG3, трансфицированных либо FLAG-меченным SOD1 дикого типа, либо SOD1, несущим мутацию G85R до и после обработки MG132, показаны на (B) и ( C) соответственно.Справа от каждого набора также показаны изображения фазового контраста. Звездочки указывают на нетрансфицированные клетки. Обратите внимание, что заметное образование агрегатов происходит только в мутантных SOD1-экспрессирующих клетках и процессах после обработки MG132. Масштабные линейки = 25 мкм.
Зависимая от микротрубочек фракция вызванного деполяризацией высвобождения ACh из клеток PNG3 требует KAP3
В нейронах Drosophila моторный комплекс kinesin-2 отвечает за антероградный аксональный транспорт ChAT (18,19).Чтобы проверить, верно ли то же самое для млекопитающих, мы сначала разработали модель анализа для обнаружения вызванного деполяризацией микротрубочек-зависимого высвобождения ACh в результате транспорта ChAT. Дифференцированные клетки PNG3 метаболически метили [ 3 H] холином и измеряли вызванное деполяризацией высвобождение [ 3 H] ACh. Мы наблюдали, что клетки PNG3 высвобождали [ 3 H] ACh дозозависимым образом от KCl (фиг. 6A). Чтобы отличить высвобождение АХ «нервных окончаний» от экзоцитоза в других местах, мы исследовали высвобождение АХ с колхицином и без него, дестабилизирующим реагентом микротрубочек, который ингибирует аксональный транспорт (18).Из экспериментов по высвобождению ACh с различными пробными количествами колхицина мы решили использовать колхицин в дозе 2,5 мкм для лучшей дифференциации микротрубочко-зависимого высвобождения, представляющего высвобождение из терминальных дифференцированных клеточных процессов PNG3. Обработка клеток этой концентрацией превращала почти 20% тубулина из нитчатой в свободную форму в клетках PNG3 (дополнительный материал, рис. S3A). Затем, чтобы проанализировать участие KAP3 / кинезин-2 в транспорте ChAT в нейронах млекопитающих с использованием этой системы анализа, мы исследовали эффект подавления регуляции мРНК KAP3 в дифференцированных клетках PNG3.Хотя клетки NG108-15 являются гибридом мышь-крыса, трансфекция siRNA для мышиного KAP3 в клетках PNG3 снижает уровень белка KAP3 до ~ 15% через 2 дня (дополнительный материал, рис. S3B). Мы обнаружили, что siRNA-опосредованное подавление KAP3 значительно снижает зависимую от микротрубочек фракцию высвобождения ACh из клеток PNG3 (рис. 6B). Это снижение высвобождения ACh было устранено котрансфекцией полноразмерного человеческого KAP3, на который не влияет использованная нами миРНК мыши. Однако высвобождение ACh не было восстановлено человеческим KAP3, лишенным С-концевой последовательности (513–792 аминокислоты), которая необходима для взаимодействия с KIF3A / B (26).Эти результаты предполагают, что KAP3 необходим для транспорта ChAT по микротрубочкам и последующего высвобождения ACh из нервных окончаний в нейронах млекопитающих.
Рисунок 6.
Зависимая от микротрубочек фракция вызванного деполяризацией высвобождения ACh из клеток PNG3 требует KAP3. ( A ) Дифференцированные клетки PNG3, метаболически меченные [ 3 H] холином, деполяризовали повышенным содержанием KCl в среде в течение 15 мин. Радиоактивность выделялась в среду, представляющую [ 3 H] ACh, и ее измеряли для различных концентраций KCl и колхицина, как указано.( B ) Вызванное деполяризацией высвобождение [ 3 H] ACh из дифференцированных клеток PNG3, трансфицированных только вектором, миРНК для мышиного KAP3 (mKAP3) или миРНК для mKAP3 вместе с плазмидой экспрессии для человеческого KAP3 (hKAP3) или hKAP3, лишенного C-концевые 223 аминокислоты (ΔhKAP3), экспрессионную плазмиду только для hKAP3 или экспрессионную плазмиду для ΔhKAP3 определяли с обработкой колхицином или без нее. Радиоактивность показана в процентах по отношению к радиоактивности трансфицированных вектором, необработанных колхицином клеток.Звездочка указывает на несущественные различия, рассчитанные как P > 0,05. Обратите внимание, что подавление KAP3 ингибирует зависимое от микротрубочек высвобождение ACh. ( C ) Обобщенный временной график для измерения высвобождения [ 3 H] ACh в дифференцированных клетках PNG3 с неправильной укладкой / агрегацией SOD1 и без нее. Вызванное KCl высвобождение ACh рассчитывали путем вычитания радиоактивности в S1 (среда до добавления KCl) из радиоактивности в S2 (среда после добавления KCl). ( D ) Вызванное деполяризацией высвобождение [ 3 H] ACh из дифференцированных клеток PNG3, трансфицированных только вектором, экспрессионную плазмиду для SOD1 дикого типа или G85RSOD1 определяли с обработкой MG132 или без нее.Зависимые от микротрубочек и независимые фракции высвобождения измеряли для каждого анализируемого состояния. Радиоактивность показана в процентах от радиоактивности клеток, трансфицированных вектором, необработанных колхицином. Обратите внимание, что высвобождение ACh из мутантных клеток, трансфицированных SOD1, снижалось в ответ на обработку MG132, в то время как сверхэкспрессия мутантного SOD1 per se не влияла на включение холина в клетки (дополнительный материал, рис. S4C). Звездочка указывает на незначительную разницу, рассчитанную как P > 0.05. ( E ) Вызванное деполяризацией высвобождение [ 3 H] ACh из дифференцированных клеток PNG3, трансфицированных только вектором, определяли плазмиду экспрессии для SOD1 дикого типа или G85RSOD1 с котрансфекцией экспрессионной плазмиды mKAP3 или без нее. после обработки MG132. Зависимые от микротрубочек и независимые фракции высвобождения измеряли для каждого анализируемого состояния. Радиоактивность показана в процентах от радиоактивности клеток, трансфицированных вектором, необработанных колхицином. Обратите внимание, что снижение высвобождения ACh, наблюдаемое в мутантных клетках, трансфицированных SOD1, в ответ на обработку MG132, было нормализовано экспрессией mKAP3.Звездочка указывает на незначительную разницу, рассчитанную как P > 0,05.
Рисунок 6.
Зависимая от микротрубочек фракция вызванного деполяризацией высвобождения ACh из клеток PNG3 требует KAP3. ( A ) Дифференцированные клетки PNG3, метаболически меченные [ 3 H] холином, деполяризовали повышенным содержанием KCl в среде в течение 15 мин. Радиоактивность выделялась в среду, представляющую [ 3 H] ACh, и ее измеряли для различных концентраций KCl и колхицина, как указано.( B ) Вызванное деполяризацией высвобождение [ 3 H] ACh из дифференцированных клеток PNG3, трансфицированных только вектором, миРНК для мышиного KAP3 (mKAP3) или миРНК для mKAP3 вместе с плазмидой экспрессии для человеческого KAP3 (hKAP3) или hKAP3, лишенного C-концевые 223 аминокислоты (ΔhKAP3), экспрессионную плазмиду только для hKAP3 или экспрессионную плазмиду для ΔhKAP3 определяли с обработкой колхицином или без нее. Радиоактивность показана в процентах по отношению к радиоактивности трансфицированных вектором, необработанных колхицином клеток.Звездочка указывает на несущественные различия, рассчитанные как P > 0,05. Обратите внимание, что подавление KAP3 ингибирует зависимое от микротрубочек высвобождение ACh. ( C ) Обобщенный временной график для измерения высвобождения [ 3 H] ACh в дифференцированных клетках PNG3 с неправильной укладкой / агрегацией SOD1 и без нее. Вызванное KCl высвобождение ACh рассчитывали путем вычитания радиоактивности в S1 (среда до добавления KCl) из радиоактивности в S2 (среда после добавления KCl). ( D ) Вызванное деполяризацией высвобождение [ 3 H] ACh из дифференцированных клеток PNG3, трансфицированных только вектором, экспрессионную плазмиду для SOD1 дикого типа или G85RSOD1 определяли с обработкой MG132 или без нее.Зависимые от микротрубочек и независимые фракции высвобождения измеряли для каждого анализируемого состояния. Радиоактивность показана в процентах от радиоактивности клеток, трансфицированных вектором, необработанных колхицином. Обратите внимание, что высвобождение ACh из мутантных клеток, трансфицированных SOD1, снижалось в ответ на обработку MG132, в то время как сверхэкспрессия мутантного SOD1 per se не влияла на включение холина в клетки (дополнительный материал, рис. S4C). Звездочка указывает на незначительную разницу, рассчитанную как P > 0.05. ( E ) Вызванное деполяризацией высвобождение [ 3 H] ACh из дифференцированных клеток PNG3, трансфицированных только вектором, определяли плазмиду экспрессии для SOD1 дикого типа или G85RSOD1 с котрансфекцией экспрессионной плазмиды mKAP3 или без нее. после обработки MG132. Зависимые от микротрубочек и независимые фракции высвобождения измеряли для каждого анализируемого состояния. Радиоактивность показана в процентах от радиоактивности клеток, трансфицированных вектором, необработанных колхицином. Обратите внимание, что снижение высвобождения ACh, наблюдаемое в мутантных клетках, трансфицированных SOD1, в ответ на обработку MG132, было нормализовано экспрессией mKAP3.Звездочка указывает на незначительную разницу, рассчитанную как P > 0,05.
Как описано выше, мы наблюдали снижение аксонального транспорта ChAT у мышей SOD1 G93A -Tg. Чтобы проверить, ингибирует ли неправильно свернутый SOD1 транспорт ChAT также в этой модельной системе FALS в культуре, мы измерили микротрубочко-зависимое высвобождение ACh из дифференцированных клеток PNG3 после индуцированного продуцирования неправильно свернутых видов SOD1 (процедура суммирована на фиг. 6C). Мы обнаружили, что индукция неправильного сворачивания SOD1 обработкой MG132 приводит к значительному снижению микротрубочко-зависимого высвобождения ACh из мутантных SOD1-экспрессирующих PNG3, но не из SOD1-экспрессирующих клеток дикого типа (рис.6D, звездочка). Сама обработка MG132 приводит к усилению регуляции как микротрубочко-зависимого, так и независимого высвобождения ACh (фиг. 6D и дополнительный материал, фиг. S4A). Однако это вызванное MG132 увеличение высвобождения ACh предположительно было вызвано повышенными уровнями экспрессии белков, необходимых для синтеза / высвобождения ACh, и не зависело от эффекта, индуцированного неправильно свернутым SOD1 (дополнительный материал, фиг. S4B и C). Эти результаты предполагают, что неправильно свернутый SOD1 ингибирует транспорт ChAT в этой модельной системе FALS в культуре.
Наблюдения на мышах SOD1 G93A -Tg, а также в описанной нами клеточной модельной системе FALS, дополнительно предполагают, что неправильно свернутый SOD1 вызывает снижение или истощение функционального комплекса кинезин-2 через его ассоциацию. с KAP3. Это повышает вероятность того, что вызванное неправильной укладкой SOD1 снижение транспорта ChAT может быть нормализовано путем добавления в мотонейроны KAP3. Чтобы доказать эту гипотезу с использованием системы модели культуры FALS, мы исследовали микротрубочко-зависимое высвобождение ACh из дифференцированных клеток PNG3, сверхэкспрессирующих SOD1 дикого типа или мутантный SOD1 вместе с KAP3.Сверхэкспрессия KAP3 не изменяла высвобождение ACh из обработанных MG132 клеток PNG, трансфицированных пустым вектором или SOD1 дикого типа. Однако сверхэкспрессия KAP3 способна нормализовать снижение высвобождения ACh в условиях, которые вызывают неправильную укладку мутантного SOD1 (фиг. 6E). Эти результаты убедительно подтверждают, что индуцированное неправильной укладкой SOD1 снижение транспорта ChAT и высвобождение ACh опосредуется снижением функциональной молекулы KAP3 из-за ее ассоциации с неправильно свернутой SOD1. Эти результаты могут также предполагать, что нормализация функционального уровня KAP3 в двигательных нейронах может иметь терапевтический эффект на патогенез FALS.
KAP3 был совместно локализован с мутантными агрегатами SOD1 в LBHI в моторных нейронах спинного мозга пациентов с FALS
Как описано, мы выполнили анализ индуцированного неправильно свернутой SOD1 снижения транспорта ChAT в культивируемых клетках в культивируемых клетках, а также в модели на мышах. Чтобы понять значение этого механизма в человеческих FALS, мы выполнили иммуногистохимию на срезах спинного мозга человеческих SOD1-связанных случаев FALS, чтобы изучить совместную локализацию KAP3 и SOD1.Невропатологические фенотипы четырех проанализированных нами пациентов, которые были членами двух разных семей (27–29), суммированы в таблице на рисунке 7. У этих пациентов часто развивались гиалиновые включения, похожие на тельца Леви (LBHI), которые представляют собой белковые агрегаты. содержащие мутантный / дикого типа SOD1 и одно из наиболее характерных цитопатологических изменений в спинномозговых мотонейронах мутантного sod1 -связанного FALS (28). Иммуногистохимический анализ показал, что большинство LBHI в моторных нейронах спинного мозга были положительными как для KAP3, так и для SOD1 (стрелки на рис.7). Мы подтвердили, что сигнал KAP3 в LBHI отсутствовал, когда антитело против KAP3 предварительно инкубировали с избытком синтетических пептидов антигена KAP3 (данные не показаны). Эти наблюдения продемонстрировали, что взаимодействие KAP3-SOD1 и последующая коагрегация часто происходит в случаях FALS, связанных с SOD1 человека.
Рисунок 7.
KAP3 был совместно локализован с агрегатами SOD1 в LBHI в спинномозговых двигательных нейронах из случаев FALS человека. Последовательные срезы LBHI-содержащих моторных нейронов спинного мозга от пациентов с FALS, обладающих мутациями sod1 и , иммуноокрашивали антителами против KAP3 и против SOD1.Клинико-патологические характеристики пациентов с FALS сведены в таблицу. Мутация D125X представляет собой делецию двух нуклеотидов в кодоне 126, которая приводит к усечению пяти остатков ниже по течению. Совместная локализация KAP3 и SOD1 в LBHI очевидна (стрелки). Изображения представляют 30 LBHI, наблюдаемых в каждом случае, все из которых показывают совместную локализацию SOD1 и KAP3. Масштабные полосы = 25 мкм для изображений верхнего ряда, 50 мкм для остальных.
Рисунок 7.
KAP3 был совместно локализован с агрегатами SOD1 в LBHI в спинномозговых мотонейронах из случаев FALS человека.Последовательные срезы LBHI-содержащих моторных нейронов спинного мозга от пациентов с FALS, обладающих мутациями sod1 и , иммуноокрашивали антителами против KAP3 и против SOD1. Клинико-патологические характеристики пациентов с FALS сведены в таблицу. Мутация D125X представляет собой делецию двух нуклеотидов в кодоне 126, которая приводит к усечению пяти остатков ниже по течению. Совместная локализация KAP3 и SOD1 в LBHI очевидна (стрелки). Изображения представляют 30 LBHI, наблюдаемых в каждом случае, все из которых показывают совместную локализацию SOD1 и KAP3.Масштабные полосы = 25 мкм для изображений верхнего ряда, 50 мкм для остальных.
ОБСУЖДЕНИЕ
В этой статье мы представили новый патологический механизм, с помощью которого неправильно свернутый SOD1 может вызывать дисфункцию нейронов при FALS. Значительная часть неправильно свернутой SOD1 находится в моторных аксонах до начала заболевания у мышей модели SOD1 G93A -Tg FALS. Мы показали, что неправильно свернутые виды SOD1 избирательно связываются с KAP3 (рис. 2B и C) и что это связывание может быть причиной снижения функциональной молекулы KAP3, необходимой для кинезин-2-зависимого транспорта в моторных аксонах (рис.3А и В). Моторный комплекс kinesin-2 особенно важен, потому что он необходим для транспорта ChAT (19). Снижение экспрессии ChAT и, как следствие, уменьшение высвобождения ACh из окончаний двигательных нервов может привести к дисфункции моторных синапсов на окончаниях аксонов (30,31). Используя недавно разработанную модель культуры клеток FALS, мы показали, что высвобождение ACh из нервных окончаний было снижено предположительно из-за снижения транспорта ChAT в результате секвестрации KAP3 неправильно свернутыми видами SOD1 (рис. 5A, C и 6D).Патологический механизм, который мы предлагаем на основе наших данных, схематически представлен на Рисунке 8.
Рисунок 8.
Схематическая диаграмма токсичности мутантной SOD1 и нарушения аксонального транспорта ChAT. В здоровых двигательных нейронах ( A ) ChAT непрерывно транспортируется двигателем кинезина-2, включающим KIF3A, KIF3B / C и KAP3, к нервно-мышечному соединению, где ACh функционирует как нейротрансмиттер. ACh высвобождается в синаптическую щель при стимуляции и быстро разлагается на холин и уксусную кислоту под действием AChE.Холин захватывается нервными окончаниями транспортерами холина, а затем ChAT синтезирует ACh из холина и ацетил-КоА. Реакция, катализируемая ChAT, является лимитирующей стадией для доставки ACh в нервные окончания. В двигательных нейронах, экспрессирующих мутантный SOD1 ( B ), мутантные белки SOD1 были преобразованы в неправильно свернутые формы в результате увеличения генерации ROS и / или снижения протеасомной активности. Некоторые неправильно свернутые виды предпочтительно связываются с KAP3 и ингибируют связывание ChAT с KAP3, тем самым вызывая снижение транспорта ChAT.Это снижение ChAT в нервных окончаниях может представлять собой механизм, вызывающий двигательную дисфункцию.
Рисунок 8.
Схематическая диаграмма токсичности мутанта SOD1 и нарушение аксонального транспорта ChAT. В здоровых двигательных нейронах ( A ) ChAT непрерывно транспортируется двигателем кинезина-2, включающим KIF3A, KIF3B / C и KAP3, к нервно-мышечному соединению, где ACh функционирует как нейротрансмиттер. ACh высвобождается в синаптическую щель при стимуляции и быстро разлагается на холин и уксусную кислоту под действием AChE.Холин захватывается нервными окончаниями транспортерами холина, а затем ChAT синтезирует ACh из холина и ацетил-КоА. Реакция, катализируемая ChAT, является лимитирующей стадией для доставки ACh в нервные окончания. В двигательных нейронах, экспрессирующих мутантный SOD1 ( B ), мутантные белки SOD1 были преобразованы в неправильно свернутые формы в результате увеличения генерации ROS и / или снижения протеасомной активности. Некоторые неправильно свернутые виды предпочтительно связываются с KAP3 и ингибируют связывание ChAT с KAP3, тем самым вызывая снижение транспорта ChAT.Это снижение ChAT в нервных окончаниях может представлять собой механизм, вызывающий двигательную дисфункцию.
БАС-подобная патология двигательных нейронов наблюдается у трансгенных мышей, экспрессирующих усеченный на С-конце SOD1 (L126Z), у которых мутантная экспрессия SOD1 не наблюдалась четко в седалищном нерве (32). Это указывает на то, что ассоциация KAP3-неправильно свернутого SOD1 формируется в основном в телах клеток двигательных нейронов. Действительно, мы обнаружили ассоциацию неправильно свернутой KAP3 SOD1 в спинном мозге (рис. 2С) и вентральном белом веществе (рис.2A и B) мышей SOD1 G93A -Tg до начала заболевания, но не смогли обнаружить его в дистальном отделе седалищного нерва даже на конечной стадии (данные не показаны). Эти находки предполагают, что даже если неправильно свернутый SOD1 ассоциируется с молекулярными моторами, он не может перемещаться на большие расстояния, предположительно потому, что он не является физиологическим грузом.
Мы подозреваем, что неправильно свернутая ассоциация SOD1-KAP3 является одним из ранних событий в патогенезе FALS. Доказательствами, подтверждающими эту возможность, является то, что ассоциация SOD1-KAP3 становится обнаруживаемой примерно в возрасте 6 месяцев у мышей SOD1 G93A -Tg, на 2 месяца раньше начала заболевания (рис.2C), подтверждая, что ассоциация SOD1-KAP3 составляет раннее событие в двигательных нейронах FALS. Кроме того, наш анализ иммунопреципитации / иммуноблоттинга показывает, что ассоциация SOD1-KAP3 всегда выявляла относительно низкомолекулярную неправильно свернутую SOD1 (-50 кДа) среди всех неправильно свернутых продуктов размером до сотен килодальтон (Рис. 2B, C и 5A). Преимущественная ассоциация KAP3 для низкомолекулярных видов, но не с большими преципитатами, указывает на то, что ассоциация SOD1-KAP3 составляет часть начальных событий в патогенезе FALS.Предыдущие сообщения об избирательном увеличении транскрипции KAP3 в поясничном отделе спинного мозга у мышей SOD1 G86R -Tg в качестве раннего события до начала заболевания (33) также подтверждают нашу гипотезу, поскольку это увеличение можно рассматривать как компенсацию Секвестрация KAP3 ассоциацией SOD1 – KAP3.
Предыдущие сообщения показывают, что скрещивание мутантных мышей SOD1-Tg с мышами, мутантными по тяжелой цепи динеина ( loa или cra ), задерживает начало заболевания и увеличивает выживаемость (34,35).Этот результат является неожиданным, потому что мутации в тяжелой цепи динеина, субъединице моторного комплекса динеина, или p150 Glued , субъединице динактина (активатор динеина), вызывают дисфункцию и дегенерацию моторных нейронов спинного мозга (15, 16). Это предполагает возможную механистическую связь между нарушением аксонального транспорта у мутантных мышей SOD1 -Tg и у мышей с мутантными динеиновыми тяжелыми цепями. Здесь мы продемонстрировали, что секвестрация KAP3 с помощью неправильно свернутой SOD1 является механизмом избирательного ингибирования аксонального транспорта, наблюдаемого у мутантных мышей SOD1-Tg.Предыдущие сообщения о транспорте меланофора в меланосомах Xenopus показали, что динактин служит активатором кинезина-2 посредством прямого связывания p150 Glued с KAP3 и регулирует внутриклеточное направление транспорта меланосом путем связывания либо с динеином (ретроградным), либо с KAP3 ( антероградный) (36). Динеин и KAP3 имеют один и тот же сайт связывания на p150 Glued , поэтому p150 Glued связывается только с одним из них за раз и тем самым активирует транспорт только в одном направлении.Если аналогичная регуляция динактином играет роль в нейронах млекопитающих, мутации loa и cra могут увеличить вероятность связывания p150 Glued с KAP3, поскольку эти мутации способствуют разборке субъединицы и повышают уровень динактина, не связанного с динеином (15). . По этому механизму мутации тяжелой цепи динеина могут приводить к нормализации опосредованного кинезином-2 транспорта ChAT, ингибируемого неправильно свернутыми видами SOD1. Действительно, в нашем анализе вентрального белого вещества мышей SOD1 G93A -Tg с использованием центрифугирования в градиенте плотности по Найкоденцу небольшая фракция p150 Glued мигрировала совместно с неправильно свернутыми видами SOD1 и KAP3 (рис.2А). Эта популяция p150 Glued также осаждалась совместно с KAP3 (данные не показаны). Этот результат может отражать участие p150 Glued в неправильном сворачивании и агрегации KAP3 / SOD1 посредством его взаимодействия с KAP3.
Недавнее исследование предоставило интересные доказательства того, что транскрипция всех членов семейства KIF3 избирательно снижается в моторной коре головного мозга пациентов со спорадическим БАС среди 13 видов кинезина и связанных с кинезином белков (37). Нарушение нервно-мышечной передачи было обнаружено у пациентов с ранним спорадическим БАС (38), и снижение количества высвобождаемого АХ считалось основной причиной дегенерации моторного терминала в модели наследственной спинальной мышечной атрофии у собак (30,39).Возможно, что дисфункция кинезина-2 и последующее ингибирование транспорта ChAT могут быть одним из общих путей, ведущих к дисфункции, специфичной для двигательных нейронов, как в спорадических, так и в семейных случаях БАС.
Грузы и адапторные молекулы для кинезина-2 изучены недостаточно. Фактор диссоциации малых GTP-связывающих белков SmgGDS (40), фодрин (41), опухолевый супрессор аденоматозного полипоза coli (42), N-кадгерин (43), ChAT и AChE (18) были описаны как молекулы, которые связываются с KAP3.Наше открытие, что неправильно свернутые виды SOD1 секвестрируют KAP3, повышает вероятность того, что транспорт этих молекул также может быть нарушен в мутантных SOD1-связанных FALS вместе со сниженным транспортом ChAT. В самом деле, мы наблюдали небольшое снижение транспорта N-кадгерина в моторных аксонах от пре-симптоматических мышей SOD1 G93A -Tg (Supplementary Material, Fig. S5). Экспрессия и функция этих молекул в моторных нейронах должны быть изучены, чтобы выяснить весь механизм.
МАТЕРИАЛЫ И МЕТОДЫ
Все данные на рисунках 1–6 являются репрезентативными для трех мышей на группу или трех независимых экспериментов. Статистическую значимость оценивали с помощью ANOVA с последующим тестом Фишера. Планки погрешностей на графиках представляют собой стандартные отклонения. Сигналы в иммуноблотах определяли с помощью ECL (GE Healthcare) и количественно оценивали с помощью программного обеспечения NIH image (1.61J).
Реагенты
Все четыре типа (WT, G93A, G85R и A4V) кДНК SOD1 человека с меткой FLAG на С-конце были клонированы в pcDNA3 (44).Полноразмерный мышиной (ID клона изображения: 5698182) и человеческий (4829354) KAP3 были приобретены из банка клонов изображений и клонированы в pcDNA3 и pCMV-Tag1, соответственно. Целостность каждого клона проверяли анализом нуклеотидной последовательности. SiRNA, предназначенная для подавления мышиного KAP3 (M-047278-00-0010), была приобретена у Dharmacon Research. Первичные антитела, используемые для иммуноблотов, были следующими: анти-SOD1 (Stressgen, SOD-100 и Calbiochem, 574597), кинезин C2 (Affinity BioReagents) и CRMP-2 (Immuno-Biological Laboratories).Антитела против ChAT (AB144P), KHC (MAB1614), KLC (MAB1617), синаптофизина (MAB5258-20UG), актина (MAB1501), нейрофиламента-M (AB1987) и MBP (основной белок миелина, AB9046) были приобретены у Chemicon. . Антитела против KAP3, KIF3A, KIF3B, p150 Glued , p50 динамитин, динеин IC, KIF2 и Rab3 были приобретены в BD PharMingen / Transduction Laboratories. Первичные антитела, используемые для иммунопреципитации и иммуноокрашивания, представляли собой антитела против SOD1 (Stressgen, SOD-100), ChAT (Chemicon, AB144P) и FLAG (Sigma, F3165).Антитело KAP3 получали путем иммунизации кроликов пептидом, содержащим 1–14 аминокислот мышиного KAP3, которые консервативны в KAP3 человека, с последующей аффинной очисткой с использованием колонки, конъюгированной с эпитопом.
Животные
Мы использовали G1L линию трансгенных мышей, несущих G93A-мутантный человеческий ген SOD1 (B6SJL- TgN ( SOD1-G93A ) 1Gur дл ), приобретенных в лабораториях Джексона. Мыши, несущие человеческий ген SOD1 дикого типа (B6.Cg- Tg ( SOD1 ) 2Gur / J) был подарком доктора Каваматы из Киотского университета. Также использовали мышей C57BL / 6J. Трансгенных мышей подвергали обратному скрещиванию с C57BL / 6J в течение более 14 поколений. Все данные были получены на самцах мышей. Все эксперименты на животных были одобрены Комитетом по уходу за животными Национального центра неврологии и психиатрии (NCNP).
Получение суботрезков спинного мозга мыши с использованием техники LMD
Замороженные срезы спинного мозга мыши получали без фиксации и обрабатывали с помощью AS LMD (Leica), как описано ранее (17).Пять секций были объединены для каждого местоположения образца для последующих анализов.
Выделение неправильно свернутых видов SOD1 из вентрального белого вещества спинного мозга
У указанных мышей иссекали спинной мозг, и вентральную половину белого вещества на уровне L1-S3 тщательно анализировали под микроскопом. Вскрытые образцы гомогенизировали в 300 мкл буфера для гомогенизации Nz [20 мМ HEPES, pH 7,4, 120 мМ NaCl, 2 мМ ЭДТА, pH 8,0, 0,25% NP-40 и полный коктейль ингибиторов протеазы (Roche)] с последующей короткой обработкой ультразвуком. .Лизаты добавляли к 3,2 мл линейного градиента плотности Nycodenz в 10 мМ HEPES, pH 7,4 и 1 мМ EDTA, pH 8,0 (начальная концентрация Nycodenz составляла 30%). После центрифугирования при 87 479 g в течение 2 часов при 4 ° C в Optima MAX-E с использованием ротора MLS 50 (Beckman Coulter) собирали 160 мкл аликвоты сверху вниз, всего 22 фракции. . Двадцать микролитров каждой фракции собирали и анализировали с помощью иммуноблоттинга.
Иммунопреципитация
Аффинно-очищенные антитела против KAP3 и против SOD1 были захвачены с помощью Dynabeads Protein G (Dynal Biotech) и сшиты с использованием диметилпимелимидата в соответствии с протоколом производителя.Для иммунопреципитации из фракций, полученных центрифугированием в градиенте плотности Nycodenz, 30 мкл каждой фракции смешивали с конъюгированными с антителами Dynabeads в 20 мМ HEPES, pH 7,4, 120 мМ NaCl, 2 мМ EDTA и полной смеси ингибиторов протеазы. Смесь инкубировали в течение ночи при 4 ° C на качающейся платформе с последующей промывкой PBS, содержащим 0,05% Tween-20. Целевые белки элюировали 30 мкл 0,1 м лимонной кислоты (pH 2,0) в течение 2 мин, и половина элюатов была проанализирована.Для иммунопреципитации из спинного мозга или клеток PNG3, трансфицированных SOD1, ткани или клетки лизировали в буфере TN-T (50 мМ Трис-HCl, pH 7,6, 1% Triton X-100, 150 мМ NaCl), содержащем полный коктейль ингибиторов протеаз и подвергали центрифугированию при 5000 g в течение 10 мин при 4 ° C. Супернатанты инкубировали с конъюгированными с антителами Protein G Dynabeads и анализировали, как описано выше.
Получение транспортируемых компонентов из брюшного конского хвоста
Фракцию, обогащенную транспортируемыми компонентами моторных аксонов, получали из вентральных корешков хвостового спинного мозга, как описано (41), со следующими модификациями.Вентральный конский хвост дважды измельчали в 400 мкл буфера IM-D с добавлением 1 мМ АТФ и коктейлем ингибиторов протеазы (Complete; Roche) в течение 10 мин на льду. Затем их подвергали центрифугированию при 5000 g в течение 10 минут при 4 ° C. В полученных супернатантах измеряли концентрацию белка и 5 мкг каждого подвергали SDS / PAGE для оценки содержания кинезина-2. Для анализа статуса кинезина-2 фракции транспортируемых везикул, полученные от четырех мышей, подвергали обработке 4.4 мл ступенчатого градиентного центрифугирования сахарозы от 0,2 до 2%. После центрифугирования в роторе SW55 Ti (Beckman Coulter) при 50000 об / мин в течение 18 часов при 4 ° C 400 мкл каждой фракции собирали из верхней части пробирки, всего 12 фракций, и 20 мкл каждой фракции анализировали с помощью SDS. –PAGE / иммуноблоты.
Культура клеток и трансфекция
клеток NG108-15 (любезный подарок доктора Акадзавы из Токийского медицинского и стоматологического университета), гибридной клеточной линии клеток нейробластомы N18TG2 мыши и клеток глиомы C6-BU-1 крысы, культивировали в среде Игла, модифицированной Дульбекко (DMEM) ( Sigma, D5796), содержащий 10% фетальной телячьей сыворотки и 2% добавку HAT (GIBCO).Дифференцировку PNG3, субклона NG108-15, выделенного из отдельных клеток, индуцировали 1 мм дибутирил-цАМФ (Nacalai tesque) и 0,2 мкм дексаметазона (MB Biomedicals) в течение 3 дней. Трансфекцию плазмид в PNG3 выполняли с использованием Optifect или Lipofectamine 2000 (Invitrogen Life Technologies) в соответствии с протоколом производителя. Среду для культивирования меняли каждые 20–24 ч, если не указано иное. Трансфекцию миРНК и котрансфекцию миРНК плазмидами проводили в соответствии с протоколами производителя с использованием реагента для трансфекции 2 DharmaFECT (Dharmacon) и реагента для трансфекции DharmaEFCT Duo соответственно.Для индукции неправильной укладки / агрегации SOD1 клетки обрабатывали 6,5 мкм MG132 (ингибитор протеасом) через 56 часов после трансфекции, а затем обрабатывали 2,5 мм перекисью водорода в течение 2 часов через 72 часа после трансфекции. Затем клетки лизировали буфером TN-T плюс смесью полного ингибитора протеазы и подвергали центрифугированию при 10 000 g в течение 10 мин при 4 ° C для разделения на растворимые (супернатант) и нерастворимые (осадок) фракции.
Измерение [
3 H] АЧ выпускВызванное деполяризацией высвобождение [ 3 H] ACh измеряли на основании ранее описанного метода (Kumagai et al ., 1993) с несколькими модификациями. Для метаболического мечения клеток PNG3 [ 3 H] холином использовали DMEM без хлорида холина. Графики маркировки, используемых реагентов и времени инкубации показаны на рисунке 6C.
Культуральную среду заменили на DMEM, не содержащую холина, с колхицином или без него. После 2 ч инкубации в среду каждой лунки 12-луночного планшета добавляли 6,25 кБк [ 3 H] хлорид холина ([метил- 3 H] хлорид холина, 2,2 ТБк / ммоль; GE Healthcare).После 2 ч инкубации среду заменяли на буфер для измерения высвобождения ACh [20 мМ HEPES-NaOH, pH 7,4, 150 мМ NaCl, 5 мМ KCl, 1,8 мМ CaCl 2 , 0,8 мМ MgSO 4 , 25 мМ глюкозы, 25 мкм сульфата эзерина и 10 мкм гемихолиния-3 (HC-3)] и культуральные планшеты возвращали в инкубатор CO 2 . Эзерин, ингибитор холинэстеразы, был добавлен для ингибирования деградации высвобожденного ACh холинэстеразой. HC-3, ингибитор высокоаффинного транспортера холина 1, был добавлен для полного ингибирования поглощения [ 3 H] холина после смены среды.После 15 мин инкубации с эзерином и НС-3 аликвоту культуральной среды переносили в пробирку объемом 1,5 мл (среда до добавления KCl), а затем в среду добавляли KCl до конечной концентрации 50 мМ. Планшеты для культивирования быстро возвращали в инкубатор CO 2 , и через 15 мин среду переносили в новую пробирку на 1,5 мл (среда после добавления KCl). Обе среды, собранные до и после добавления KCl, центрифугировали при 10 000 g в течение 5 минут при 4 ° C для удаления мусора.Супернатанты смешивали со сцинтилляционной смесью (Ultima Gold XR, Perkin Elmer) для измерения радиоактивности [ 3 H] с использованием жидкостного сцинтилляционного спектрофотометра. Радиоактивность [ 3 H] ACh, высвобождаемого KCl, рассчитывали путем вычитания активности среды до KCl (= S1) из активности среды после добавления KCl (= S2), как показано на Фигуре 6C.
Иммуноцитохимия
клеток PNG3 фиксировали 4% параформальдегидом в PBS в течение 15 мин.После пермеабилизации 0,15% Triton X-100 в PBS в течение 10 минут клетки обрабатывали Blocking-one (Nacalai) в течение 1 часа. Затем клетки обрабатывали первичными антителами в Blocking-one при 4 ° C в течение ночи. Клетки промывали PBS. Для обнаружения клетки обрабатывали флуоресцентно-конъюгированными вторичными антителами в Blocking-one при комнатной температуре в течение 1 часа. После повторной промывки PBS клетки фиксировали с помощью Vectasheld (Vector Laboratories).
Вскрытие и иммуногистохимия
Исследования проводились на архивных, забуференных 10% фиксированных формалином, залитых парафином тканях спинного мозга, полученных при аутопсии у четырех пациентов с FALS, которые были членами двух разных семей.Согласие на вскрытие было получено от законных представителей в соответствии с требованиями местных наблюдательных комиссий. Клинико-патологические характеристики пациентов с FALS были описаны ранее (27,29). Анализ гена SOD1 показал, что члены японского семейства Oki имели делецию 2 п.н. в кодоне 126 в гене SOD1 (мутация 126 со сдвигом рамки считывания, D125X) и что члены семейства T имели замену лейцина на валин в кодоне 106 в гене SOD1 (L106V).Чтобы посмотреть на экспрессию KAP3 и SOD1 в этих образцах, аффинно очищенное кроличье анти-KAP3 антитело (0,05 мкг / мл, разведенное 1: 200 в 1% бычьем сывороточном альбумине, содержащем PBS, pH 7,4), мышиное моноклональное антитело против человеческого В качестве первичных антител использовали SOD1 (0,5 мг / мл, clone1G 2 , MBL, Aichi, Япония) и поликлональные антитела овцы против SOD1 человека (1: 20 000, Calbiochem, Дармштадт, Германия). Иммуногистохимическую реакцию проводили по стандартной методике. Сигнал антител визуализировали методом авидин-биотин-иммунопероксидазного комплекса (ABC) с использованием соответствующего набора Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA) и 3,3′-диаминобензидинтетрагидрохлорида (DAB; Dako, Glostrup, Дания) как хромоген.
ДОПОЛНИТЕЛЬНЫЙ МАТЕРИАЛ
Дополнительные материалы доступны в HMG онлайн.
ФИНАНСИРОВАНИЕ
Эта работа была поддержана исследовательскими грантами от RIKEN BSI, субсидии на научные исследования в приоритетной области (проект Advanced Brain Science Project) Министерства образования, культуры, спорта, науки и технологий, NIBIO (Программа содействия развитию Fundamental Studies in Health Sciences), грант Японского фонда нейробиологии и психического здоровья и грант Министерства здравоохранения, труда и социального обеспечения Японии.Средства на оплату открытого доступа были предоставлены за счет гранта Министерства здравоохранения, труда и социального обеспечения Японии.
БЛАГОДАРНОСТИ
Мы хотим поблагодарить сотрудников лаборатории за полезные предложения, а также Томоко Дай, Марико Сода, Юмико Симадзаки и Тадатоши Макино за их техническую поддержку.
Заявление о конфликте интересов . Ничего не объявлено.
ССЫЛКИ
1,,.БАС: болезнь мотонейронов и их ненейронных соседей
,Neuron
,2006
, т.52
(стр.39
—59
) 2,.Молекулярная биология бокового амиотрофического склероза: выводы из генетики
,Nat. Rev. Neurosci.
,2006
, т.7
(стр.710
—723
) 3,.Каким образом мутации супероксиддисмутазы 1, связанные с БАС, способствуют агрегации белка?
,Trends Biochem. Sci.
,2007
, т.32
(стр.78
—85
) 4,,.Окислительный стресс при БАС: механизм нейродегенерации и терапевтическая мишень
,Biochim. Биофиз. Acta.
,2006
, т.1762
(стр.1051
—1067
) 5,,.Высокомолекулярные комплексы мутантной супероксиддисмутазы 1: возрастное и тканеспецифическое накопление
,Neurobiol. Дис.
,2002
, т.9
(стр.139
—148
) 6,,,.Иммуногистохимическое исследование супероксиддисмутаз в спинном мозге вскрытых пациентов с боковым амиотрофическим склерозом
,Dev.Neurosci.
,1996
, т.18
(стр.492
—498
) 7,,,,,.Гистологические доказательства агрегации белков у трансгенных мышей с мутантным SOD1 и нервных тканей с боковым амиотрофическим склерозом
,Neurobiol. Дис.
,2001
, т.8
(стр.933
—941
) 8,,,,,,.Дисульфид-восстановленная супероксиддисмутаза-1 в ЦНС моделей трансгенного бокового амиотрофического склероза
,Мозг
,2006
, т.129
(стр.451
—464
) 9,,,,,,,,, и др.Изменение растворимости семейного мутанта SOD1, связанного с БАС, с прогрессированием заболевания: его модуляция протеасомой и Hsp70
,Biochem. Биофиз. Res. Commun.
,2006
, т.343
(стр.719
—730
) 10,.Транспортные системы на основе микротрубочек в нейронах: роль кинезинов и динеинов
,Annu. Rev. Neurosci.
,2000
, т.23
(стр.39
—71
) 11,.Молекулярные моторы и механизмы направленного транспорта в нейронах
,Нат. Rev. Neurosci.
,2005
, т.6
(стр.201
—214
) 12,,,.Роль аксонального транспорта в нейродегенеративных заболеваниях
,Annu. Rev. Neurosci.
,2008
, т.31
(стр.151
—173
) 13,,.Дефектный аксональный транспорт в модели бокового амиотрофического склероза у трансгенных мышей
,Nature
,1995
, vol.375
(стр.61
—64
) 14,.Замедление аксонального транспорта является очень ранним проявлением токсичности связанных с БАС мутантов SOD1 для мотонейронов
,Nat. Neurosci.
,1999
, т.2
(стр.50
—56
) 15,,,,,,,,, и др.Мутации в динеине связывают дегенерацию двигательных нейронов с дефектами ретроградного транспорта
,Science
,2003
, vol.300
(стр.808
—812
) 16,,,,,,,,, и др.Мутантный динактин при заболевании двигательных нейронов
,Nat. Genet.
,2003
, т.33
(стр.455
—456
) 17,,,,,,,,, и др.Проницаемые для кальция рецепторы AMPA способствуют неправильной укладке мутантного белка SOD1 и развитию бокового амиотрофического склероза у трансгенных мышей модели
,Hum. Мол. Genet.
,2004
, т.13
(стр.2183
—2196
) 18,,,,.Кинезин-2 дифференциально регулирует антероградный аксональный транспорт ацетилхолинэстеразы и холинацетилтрансферазы у Drosophila
,J. Neurobiol.
,2006
, т.66
(стр.378
—392
) 19,,,,,,.Кинезин-II необходим для аксонального транспорта холинацетилтрансферазы в Drosophila
,J.Cell Biol.
,1999
, т.147
(стр.507
—518
) 20.Активизация разработки с помощью гетеротримерного кинезина KIF3
,Traffic
,2000
, vol.1
(стр.29
—34
) 21,.Холин и холинергические нейроны
,Science
,1983
, т.221
(стр.614
—620
) 22,,,,,.Регулирование высвобождения ацетилхолина из гибридных клеток нейробластомы × глиомы
,Proc.Natl Acad. Sci. США
,1978
, т.75
(стр.1314
—1318
) 23,,,.Приобретение нейронных белков при дифференцировке клеток NG108-15
,Neurosci. Res.
,2000
, т.37
(стр.153
—161
) 24,,,.Образование высокомолекулярных комплексов мутантной Cu, Zn-супероксиддисмутазы на мышиной модели семейного бокового амиотрофического склероза
,Proc.Natl Acad. Sci. США
,2000
, т.97
(стр.12571
—12576
) 25,,,,.Агрегатное образование в спинном мозге мутантных трансгенных мышей SOD1 обратимо и опосредуется протеасомами
,J. Neurochem.
,2003
, т.87
(стр.851
—860
) 26,,,,.Роль белка семейства кинезин-2, KIF3, во время митоза
,J. Biol. Chem.
,2006
, т.281
(стр.4094
—4099
) 27,,,,,.Семейный амиотрофический боковой склероз с делецией двух пар оснований в супероксиддисмутазе 1: мультисистемная дегенерация гена с внутрицитоплазматическими включениями гиалина в астроцитах
,J. Neuropathol. Exp. Neurol.
,1996
, т.55
(стр.1089
—1101
) 28,,,,,,,,.Патологическая характеристика астроцитарных гиалиновых включений при семейном боковом амиотрофическом склерозе
,Am.J. Pathol.
,1997
, т.151
(стр.611
—620
) 29.Модели бокового амиотрофического склероза и невропатология человека: сходства и различия
,Acta Neuropathol.
,2008
, т.115
(стр.97
—114
) 30,,,.Пониженное содержание нервно-мышечных квантов с нормальным течением времени высвобождения синапсов и депрессией при болезни мотонейронов собак
,J. Neurophysiol.
,2002
, т.88
(стр.3305
—3314
) 31,,,,,,,,, и др.Аберрантное формирование паттерна нервно-мышечных синапсов у мышей с дефицитом холинацетилтрансферазы
,J. Neurosci.
,2003
, т.23
(стр.539
—549
) 32,,,,,,,,.Соматодендритное накопление неправильно свернутой SOD1-L126Z в двигательных нейронах опосредует дегенерацию: альфа-В-кристаллин модулирует агрегацию
,Hum.Мол. Genet.
,2005
, т.14
(стр.2335
—2347
) 33,,,,,,,.Дифференциальный скрининг мутировавших трансгенных мышей SOD1 выявляет раннюю активацию компонента быстрого аксонального транспорта в мотонейронах спинного мозга
,Neurobiol. Дис.
,2000
, т.7
(стр.274
—285
) 34,,,,,.Мутация динеина ослабляет дегенерацию двигательных нейронов у мышей SOD1 (G93A)
,Exp.Neurol.
,2006
, т.198
(стр.271
—274
) 35,,,,,,,.Мутация динеина устраняет дефекты аксонального транспорта и увеличивает продолжительность жизни мышей с БАС
,J. Cell Biol.
,2005
, т.169
(стр.561
—567
) 36,,,,,,.Динактин необходим для двунаправленного транспорта органелл
,J. Cell Biol.
,2003
, т.160
(стр.297
—301
) 37,,,,,.Дифференциальная экспрессия молекулярных моторов в моторной коре спорадического БАС
,Neurobiol. Дис.
,2007
, т.26
(стр.577
—589
) 38,,,,,,,.Нервно-мышечная передача при боковом амиотрофическом склерозе
,Мышечный нерв
,1993
, т.16
(стр.1193
—1203
) 39,,,,,,.Нарушение функциональных двигательных единиц предшествует нервно-мышечной дегенерации при заболевании двигательных нейронов собак
,Ann.Neurol.
,2000
, т.47
(стр.596
—605
) 40,,,,,,.SMAP, Smg GDS-ассоциированный белок, имеющий повторы плеч и фосфорилированный тирозинкиназой Src
,J. Biol. Chem.
,1996
, т.271
(стр.27013
—27017
) 41,,,,,.Кинезиновый белок суперсемейства 3 (KIF3) мотор транспортирует фодрин-ассоциированные везикулы, важные для построения нейритов
,J.Cell Biol.
,2000
, т.148
(стр.1255
—1265
) 42,,,,,,.Идентификация связи между опухолевым супрессором APC и суперсемейством кинезинов
,Nat. Cell Biol.
,2002
, т.4
(стр.323
—327
) 43,,,,,,,.Двигатель KIF3 транспортирует N-кадгерин и организует развивающийся нейроэпителий
,Nat. Cell Biol.
,2005
, т.7
(стр.474
—482
) 44,,,.Протеасомное ингибирование неправильно свернутой мутантной супероксиддисмутазой 1 вызывает селективную гибель мотонейронов при семейном боковом амиотрофическом склерозе
,J. Neurochem.
,2002
, т.83
(стр.1030
—1042
) 45,,.Увеличение образования нейритов и высвобождения ацетилхолина путем трансфекции кДНК ассоциированного с ростом белка-43 в клетки NG108-15
,J.Neurochem.
,1993
, т.61
(стр.526
—532
)Заметки автора
© 2008 Автор (ы).
Это статья в открытом доступе, распространяемая в соответствии с условиями некоммерческой лицензии Creative Commons Attribution (http://creativecommons.org/licenses/by-nc/2.0/uk/), которая разрешает неограниченное некоммерческое использование и распространение. , а также воспроизведение на любом носителе при условии правильного цитирования оригинала.
.